
The Aviva big day out didn't work the past 2 years because every day was a big day out.
The casual fan who goes to 3 or 4 games a year will still to to the Christmas Euro & an interpro in the Aviva.
1
40

Jun 12
Modstrings
Classes for DNA & RNA sequences containing modified nucleotides
bioconductor.statistik.tu-do…
ModRNAString alphabet
Alphabet for the modifications used in this package are based on the compilation of RNA modifications by the Bujnicki lab (Boccaletto et al. 2018). The alphabet was modified to remove some incompatible characters
bioconductor.statistik.tu-do…
MODOMICS
modomics.genesilico.pl/
MODOMICS was linked to RNAcentral, a DB of non-coding RNA sequences, & serves as a source of modified tRNA & rRNA sequences
Many of the products of modification reactions are substrates for further reactions, & the formation of hypermodified residues occurs in complex pathways, which are displayed as graphs in the PATHWAYS section of the database
academic.oup.com/nar/article…
RNAcentral: non-coding RNA sequence database
rnacentral.org/
RNAcentral is a free, public resource that offers integrated access to a comprehensive & up-to-date set of non-coding RNA sequences provided by a collaborating group of Expert Databases representing a broad range of organisms & RNA types
academic.oup.com/nar/article…
RNAcentral Expert Databases
Currently the RNAcentral Consortium is formed by 62 Expert Databases, 52 of which have already been imported into RNAcentral
rnacentral.org/expert-databa…
RNAcentral assigns unique IDs to every distinct sequence & supports species-specific identifiers for referring to sequences in specific organisms
rnacentral.org/help#rnacentr…
The sequences are mapped to reference genomes from more than 250 species using blat. U can use a genome browser to browse all mapped sequences, view individual sequences in their genomic context, or d/l genome coordinates in several formats
rnacentral.org/help/genomic-…
We import modified nucleotides from Modomics & PDB, miRNA targets from TarBase, & Gene Ontology annotations from QuickGO. All sequences are annotated w/ Rfam models to provide warnings & additional info. The sequence & structural info is used to build Infernal covariance models, which can be used to find new instances of RNA families & annotate genomes w/non-coding RNAs
rnacentral.org/help/qc
We also provide secondary structure diagrams for a wide range of RNA types
rnacentral.org/help/secondar…
HMMER - biological sequence analysis using profile hidden Markov models (HMMs)
HMMER searches bio sequence databases for homologous sequences, using either single sequences or multiple sequence alignments as queries. HMMER implements a tech called "profile hidden Markov models"
github.com/EddyRivasLab/hmme…
HMMER is often used together w/ a profile database, such as Pfam (pfam.xfam.org/) or many of the databases that participate in Interpro (ebi.ac.uk/interpro/)
HMMER can also work w/ query sequences, not just profiles, just like BLAST. For example, u can search a protein query sequence against a database w/ phmmer, or do an iterative search w/ jackhmmer
HMMER is designed to detect remote homologs as sensitively as possible, relying on the strength of its underlying probability models. In the past, this strength came @ significant computational expense, but as of the new HMMER3 project, HMMER is now essentially as fast as BLAST
hmmer.org/
We believe HMMER compiles & runs on any POSIX-compliant system w/ an ANSI C99 compiler, including Mac OS/X, Linux, & any UNIX operating systems, provided it supports SSE2, ARM NEON, or big-endian Altivec/VMX vector instructions. This includes essentially all Intel/AMD compatible machines, Apple OS/X Intel or ARM machines, & some PowerPC machines
hmmer.org/documentation.html
HMMER Webserver
ebi.ac.uk/Tools/hmmer/home
HMMER User’s Guide V.3.4
eddylab.org/software/hmmer/U…
Easel - C library for bio sequence analysis
Easel, used by HMMER & Infernal, supports our work on computational analysis of bio sequences using probabilistic models. Easel aims to make similar apps more robust & easier to develop, by providing a set of reusable, documented, & well-tested functions
github.com/EddyRivasLab/ease…

Jun 12

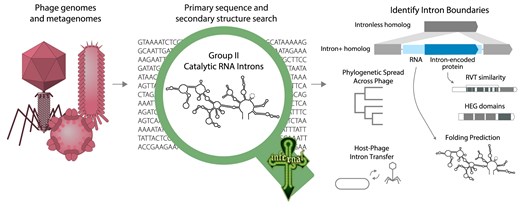
Presence of group II introns in phage genomes
Advances in RNA structure-based homology searches using covariance models has provided the ability to identify the conserved secondary structures of group II introns. Here, we discover that group II introns are widely found in phages from diverse phylogenetic backgrounds, from endosymbiont phage to jumbophage.
A general computational method for identifying homologs of a conserved RNA secondary structure and sequence consensus uses probability models called profile stochastic context-free grammars (profile SCFGs, also called covariance models) [25, 26].academic.oup.com/nar/article… academic.oup.com/nar/article…
A software package called Infernal implements profile SCFG-based search and alignment [27].academic.oup.com/bioinformat… The Rfam database of 4000 known conserved RNA structure elements is built with Infernal [28].academic.oup.com/nar/article… The input to an Infernal search is a multiple sequence alignment of the conserved RNA annotated with its consensus secondary structure. From this sequence and structure information, Infernal builds a consensus statistical model, which can then be used to search genome sequences for homologs, and to structurally align new homologs to the consensus.
Profile stochastic context-free grammar (SCFG) algorithms used to be prohibitively computationally expensive, but a set of accelerated algorithms in Infernal now allows for comprehensive searches for RNAs in large genome and metagenome datasets, including RNAs the size of catalytic introns [29].academic.oup.com/nar/article… HERE, WE USE INFERNAL TO SEARCH FOR GROUP II INTRONS IN PHAGE GENOMES.
academic.oup.com/nar/article…
Infernal ("INFERence of RNA ALignment")
Infernal ("INFERence of RNA ALignment") is for searching DNA sequence databases for RNA structure and sequence similarities. It is an implementation of a special case of profile stochastic context-free grammars called covariance models (CMs). A CM is like a sequence profile, but it scores a combination of sequence consensus and RNA secondary structure consensus, so in many cases, it is more capable of identifying RNA homologs that conserve their secondary structure more than their primary sequence.
eddylab.org/infernal/
The "cmbuild" program builds a statistical profile of your alignment. That covariance model (CM) can be used as a query in a database search to find more homologs of your RNAs (the"cmsearch" program). You can also use a covariance model (CM) of a representative alignment of your sequence family to create a larger consensus alignment of any number of RNAs (the "cmalign" program).
eddylab.org/infernal/README.…
INFERNAL User’s Guide
Sequence analysis using profiles of RNA sequence and secondary structure consensus
eddylab.org/infernal/Usergui…
Biocontainers wrapper for infernal
quay.io/repository/biocontai…
Galaxy wrapper for Infernal prediction tools
toolshed.g2.bx.psu.edu/repos…
Rfam
Rfam is a database of functional non-coding RNA families represented by multiple sequence alignments and consensus secondary structures.
rfam.org/
The Rfam database is a collection of non-coding RNA sequence families of structural RNAs, including non-coding RNA genes as well as cis-regulatory elements. Each family is represented by a multiple sequence alignment and a covariance model (CM).
docs.rfam.org/en/latest/abou…
The Rfam database of RNA families is based on the Infernal software engine. This article demonstrates how to use the Rfam database and website and includes a section on using Infernal for genome annotation.
pmc.ncbi.nlm.nih.gov/article…

4
11
22
737

Jun 11
CricketEurope News: North triumphs over South in first over-50 interpro - Cricket's veterans rolled back the years at Merrion's Anglesea Road ground as the Northern XI defeated the Southern XI by five wickets in the inaugural Over-50s Interprovincial… ift.tt/uvBliXF
2
563
Jun 10
CricketEurope News: T20 Interpro series cancelled - Cricket Ireland has today advised that the Inter-Provincial T20 Festival scheduled for Lisburn this week will not proceed as planned due to external factors. ift.tt/byWvkE7
825
Northern Cricket Union retweeted
CricketEurope News: Squads named for Ireland Over 50s interpro - The Irish Veterans Association has announced the squads for the inaugural North v South fixture, which will be played at Merrion Cricket Club on Wednesday, June 10, with play commencing… ift.tt/2rwE0bo
2
7
1,408

La CNT-SO interpro 34 relaie la pétition à l'initiative des personnels et parents d'élèves des lycées de l'Hérault pour demander plus de moyens et plus de postes !
change.org/p/des-moyens-huma…
Si vous êtes personnels ou parents d'élèves d'un lycée de l'Hérault signez ici ☝🏼 !
2
3
60
CricketEurope News: Knights and Lightning unchanged for interpro clashes - Leinster Lightning and the Northern Knights have named unchanged squads for the second of three T20 Festivals being held this week at Lisburn in the lead up to the two T20 int… ift.tt/VUnC7w8
1
600



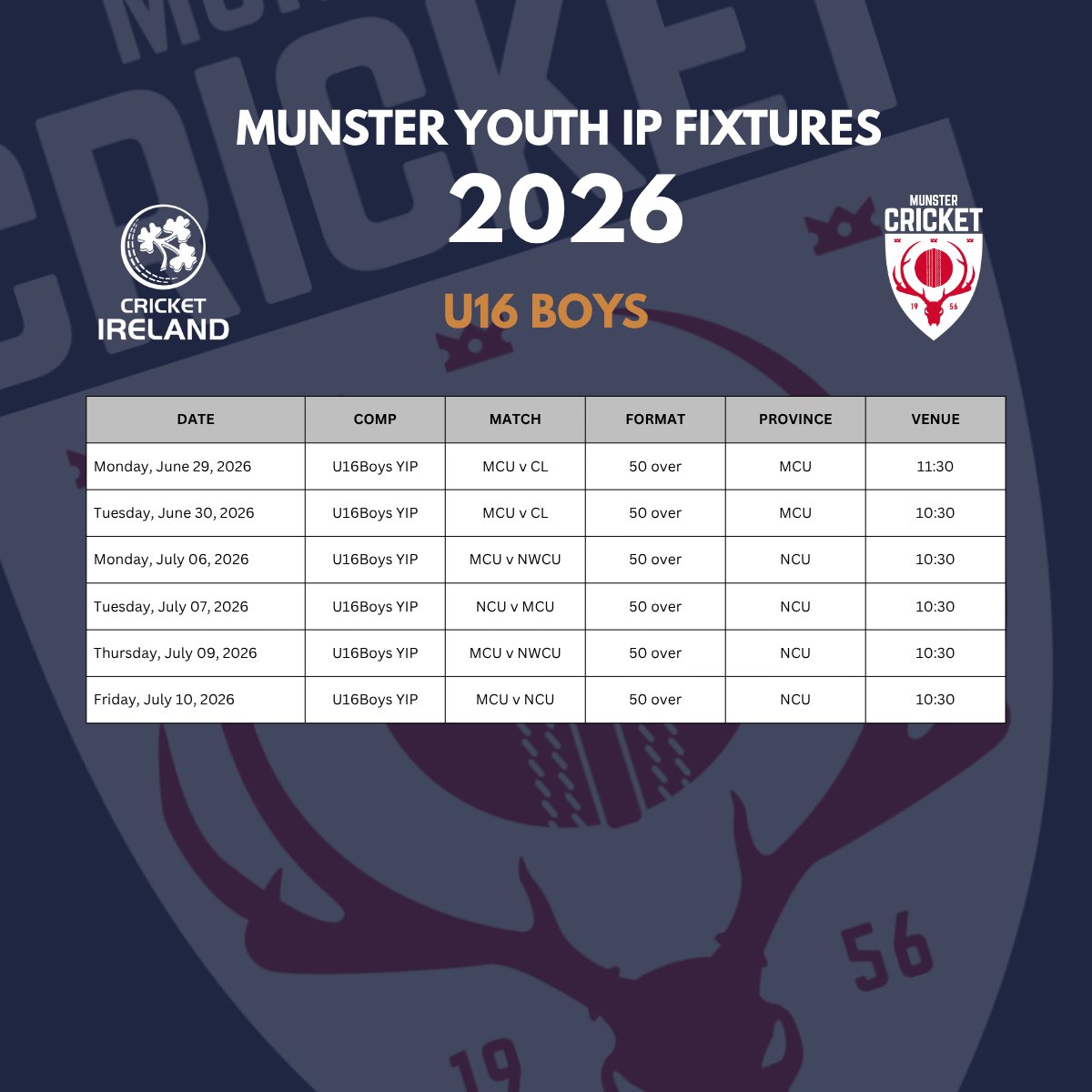
Here are your Youth Interpro and Future Series fixtures for 2026. There is some great cricket in prospect!
@cricketireland


1
2
175

May 29
Il y a deux types de grèves:
-Les corporatistes qui marchent
-Les interpro qui ne marchent pas
Pourquoi?
2
275



May 27
The 2026 Vodafone Women’s Interpro fixtures have been announced, with this year’s finals day set to take place in Galway
The opening fixture of the series will be a repeat of last years final as Leinster take on Munster

7
658

May 27
The schedule is set 🔥
Vodafone Women’s Interpro Championship fixtures confirmed ✅
Venue and kick-off details to follow.

1
18
3,014


May 25
La au moins c'est de la vraie communication pas comme celle de nos interpro française
May 24

The funniest maths in modern environmentalism.
One almond requires 12 litres of irrigated water to produce. Peer-reviewed, ScienceDirect, 2017. A glass of almond milk contains roughly 50 of them. 600 litres of water before the carton is filled.
The water comes from the San Joaquin Valley in California, which sits over one of the most over-extracted aquifers on earth. The valley floor has subsided by up to nine metres in places due to groundwater depletion. The carton is then refrigerated, sailed across the Atlantic, refrigerated again, lorried to a Manchester Tesco, and bought by someone who is concerned about the environmental impact of dairy.
Meanwhile, in Cheshire.
A British dairy cow drinks roughly 70 to 100 litres of water a day and produces around 28 litres of milk. That's about 3.5 litres of water per litre of milk. The water is rainwater that fell on her field or came from a local stream fed by the same rainwater. The rain was going to fall on the field whether the cow stood in it or not. 80% of her moisture intake comes from the grass itself, which is also rain.
She converts the grass, free of charge, into a litre of milk containing seven times the protein and four times the calcium of almond milk, and shipped roughly 18 miles to the same Tesco.
To recap.
600 litres of stolen aquifer, flown halfway round the world for nutritionally worthless beige water.
Or 3.5 litres of rain that was already falling, converted by an animal you can pet, into actual food.
The shopper picks the almond.
She has been told this is the ethical position.
The aquifer would like a word.

2
142