
There is no need for a press briefing ..
People need to know if his vehicle is not computable for E20 and above and middle class people suffer the same as almost 90% of vehicles are not compatible what is the solution ...
Why is the govt forcefully imposed ??
11

Webinar: Economic Modeling for a Polycrisis World - Introducing UNDP Computable General Equilibrium (CGE) Model for Policy Scenario Analysis sparkblue.org/event/webinar-…
1
6

They could understand exponential growth if properly incentivized. But, our political systems, all of them, were never designed for a world where multiple technologies change and converge rapidly, creating compounding effects in an environment of abundant, accessible, and computable information.
55

jejeje barsa aplastar, ni cucarachas , negreira puede ,el barsa negativo no es computable ,que diria un robotico
5

... la implantación de una jornada laboral de 35 horas, con el exceso de jornada reconocido, retribuido y computable a efectos de jubilación; una clasificación profesional justa, acorde al nivel formativo y a la responsabilidad clínica y ...
1
16

1920s-30s: The foundation gets laid.
Punch cards run the census. Vacuum tubes show up. Alan Turing publishes his paper on computable numbers in 1936.
Nobody calls it “IT” yet. But the blueprint for everything is being drawn.

1
49


This probably isn't something that people usually put out (so that everything feels magically natural), but here goes:
For the longest time, I have wondered what my personal brand should feel like to anyone reading my posts.
And to be honest, the more I think about an extravagant idea, the less authentic it feels.
So I've decided to just revolve it around who I am at my core:
I'm a developer, I love solving really hard problems and I love life.
So yeah that's it - If someone reads my posts and feels like I'm just a dev trying to solve this huge problem of making human biology computable,
I guess that'd be pretty cool.

9
307

El requisito que pide el alcalde es computable a todos los inmigrantes, sean extranjeros o no.#EnBocaDeTodos
2
41

Ni idea de cómo computan en masa salarial anual.
(Salario amortización)=
Gordon 27
Total computable anual 27
Konate 27
Bernardo Silva 26
Cucurella 28
Dumfries 19
Total computable anual 100



39
Richard Eborall retweeted

18 Nov 2022
Yes but that's not woo. He just means not Turing (nor quantum) computable. Which is merely the Mathematicians' Misconception.
1
2
8

The Joubert brothers’ shift from tech startups to luxury safaris
Malan and Philip Joubert, the South African co-founders behind OfferZen and earlier backers of several major tech startups, are now building Magic Safari (@magicsafarico), a stealth startup aimed at becoming the digital backbone of Africa’s high-end safari industry. Philip says the platform is designed to make safari planning computable, cutting what is currently a slow, error-prone process down to minutes, while helping lodges present themselves earlier in the traveller journey.
Key Takeaways
- The brothers previously founded FireID, which later incubated companies including Luno, SnapScan, Root, JourneyApps and Pondering Panda.
- Their best-known venture, OfferZen, flipped recruitment by letting companies pitch jobs to developers and has helped over 100,000 tech professionals find work.
- Magic Safari, led by Philip and built with Louw Hopley and Chris Lemmer, is already profitable in stealth and being used by some of Africa’s top luxury lists.
magicsafari.com
1
62

Hello mr --- veichles computable with e100 are coming soon u can go with them
6

Four months ago, a quantum computer simulated a protein with 303 atoms for the first time. This month, the same team simulated one with 12,635 atoms. That is a 40-fold increase, in four months.
Simulating how a drug molecule interacts with a protein at the quantum level is one of the most computationally demanding problems in chemistry. Classical computers can approximate it for small molecules, but the calculations required for biologically meaningful proteins, the kind actually involved in drug design, have been considered far beyond the reach of quantum hardware.
In late 2025, a collaboration between Cleveland Clinic, RIKEN, and IBM used quantum computing to simulate the electronic structure of Trp-cage, a 303-atom miniprotein, for the first time. It was described at the time as a milestone.
On May 5, 2026, the same team announced they had simulated two much larger protein-ligand complexes: T4-Lysozyme, at 11,608 atoms, and Trypsin, at 12,635 atoms, both modelled with bound drug-like molecules and immersed in water to replicate real biological conditions.
That is a 40-fold increase in the size of the system simulated, and a 210-fold improvement in accuracy on a key step in the calculation, achieved in four months.
The method is called quantum-centric supercomputing. Classical computers first break the massive protein-ligand complex down into smaller, computable fragments. IBM's 156-qubit Quantum Heron processors, running at facilities at both Cleveland Clinic and RIKEN, then calculate the quantum-mechanical behaviour of those fragments, using up to 94 qubits and nearly 6,000 quantum operations in certain parts of the simulation. The results are reassembled using two of the world's most powerful classical supercomputers, RIKEN's Fugaku and the University of Tokyo and University of Tsukuba's Miyabi-G.
A key technical refinement made this possible: the team restricted the most computationally expensive quantum calculations to a local sphere of 7 to 10 angstroms around specific atoms of interest, because the quantum entanglement relevant to the calculation diminishes sharply at greater distances. This let them avoid the impossible task of simulating the entire 12,635-atom system at full quantum detail.
Lead author Dr. Kenneth Merz of Cleveland Clinic described the moment: this is one of those things you dream about.
The researchers are clear about the current limitation. This method does not yet outperform the best classical computational chemistry methods for proteins. What it establishes is the trajectory: a 40-fold increase in scale and a 210-fold increase in accuracy, in four months, using a technique that is improving faster than classical methods are.
Source: IBM Quantum Blog and official press release, May 5, 2026. Cleveland Clinic, RIKEN, and IBM. Lead author: Dr. Kenneth Merz, Cleveland Clinic.
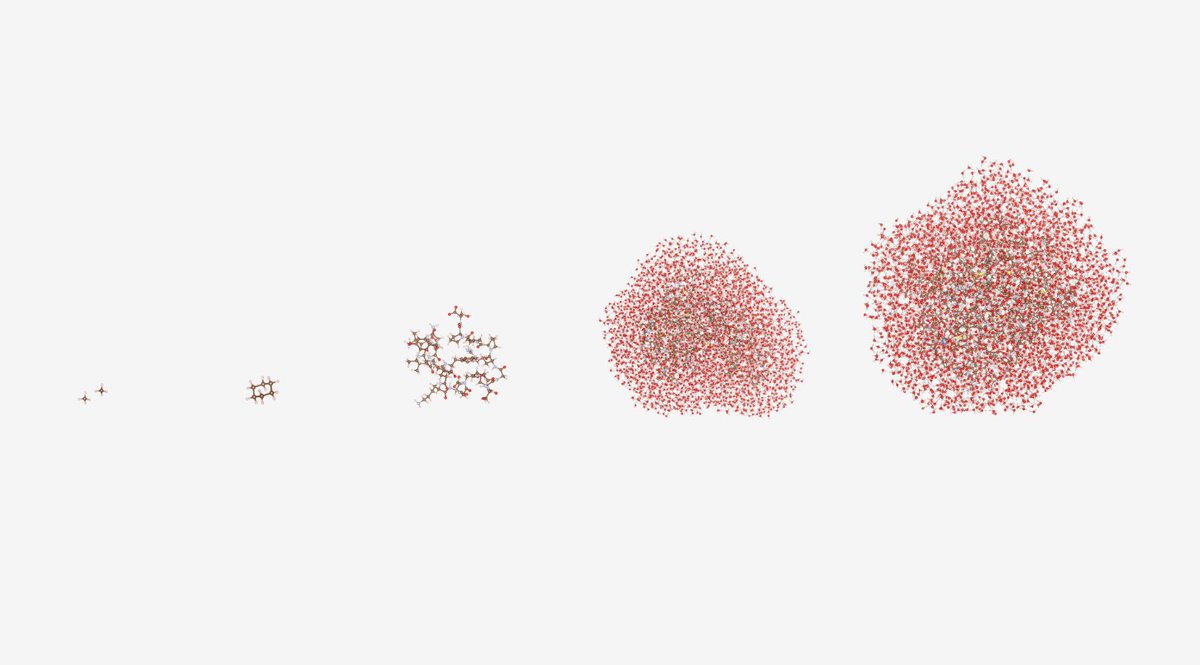
13
16
1,205