
気ままに動く
Joined April 2013
- Tweets 14,238
- Following 373
- Followers 230
- Likes 9,383
347 Photos and videos

















時期的に女子枠関係ないし、女子枠なくても理系人口増えてるならますます女子枠いらなくないですか?

1
4
24
559
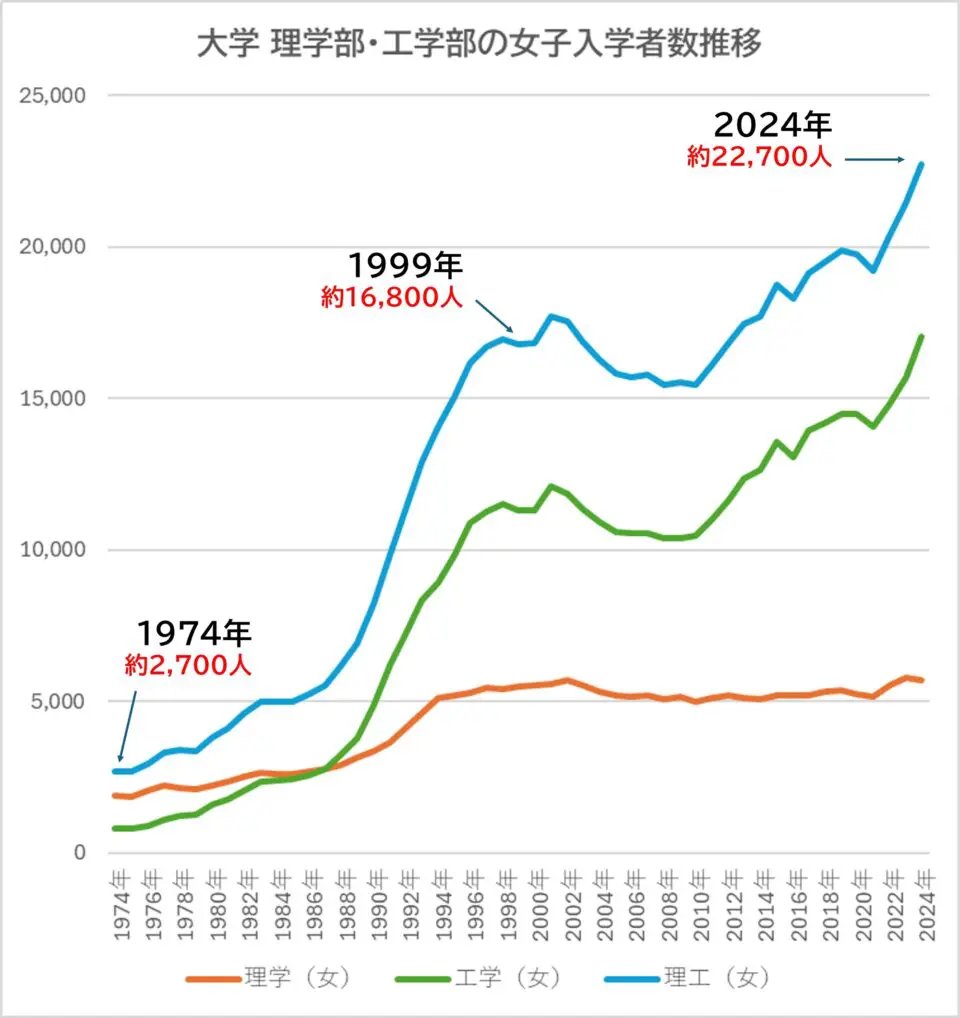
『理系の女子人口を増やすためには女子枠が必要なんです』
「女子枠作っても本来、偏差値がより低い大学に行くはずの女子高生がトップ大学に入学するだけで女子比率は増えません」
『女子比率はここ数十年増えてます(グラフ付)』
私:そしたら女子枠いらないじゃん・・・
2
61
生物系の研究したかったから迷わず生物選択で受験したけど、生物系で必修の物理の範囲ですら大学で勉強するのキツかったのだが、工学部で物理未履修で受験したら入学後についていけるのか普通に疑問です・・・
入学できても大学は地獄なんじゃないだろうか・・・

無茶苦茶だけど効果の見込める方法をご紹介します。
「工学部の入試で生物を必修にする」という方法があります。
女子の割合がめちゃくちゃ高い"生物選択者"にのみ受験資格を与え、男子の割合が多い"物理選択者"から受験資格を奪うことで、受験者の女子比率の向上が見込めます。

1
198
女性専用車両やプリクラの男子禁止、レディースデイぐらいなら「まぁ厳密には男性差別だろうけど仕方ないから我慢してあげるか」の精神でスルーできたんだが、最近は進学や就職みたいな重大イベントでも平気で差別制度が作られるから流石にスルーできなくなってきた感じがします。
Jun 12

普通に生活してると「男女論の方からやってくる」んですよね。逃げられない。受験に就活、昇進...独立しようにも創業融資...
29
1,043
5,807
279,779
ちなみに私は男だけど女性専用車両は残して良いと思ってます
まぁXだと過激な意見も多くてムッとする時もあるけど、あれで安心する女性も確かにいると思うので社会貢献で協力したい気持ちです
プリクラは私が学生の時は友達と普通に撮ってたので今の学生のためにもなんとかしてあげたいのが本音です
2
5
29
4,203
Ph.D. dolphin retweeted

Jun 12
ジンベエザメ、6年間「寄生生物を採取した研究者」を掃除魚だ思い込む
nazology.kusuguru.co.jp/arch…
豪UWAがジンベエザメの皮膚に寄生したカイアシ類の採取調査を続けたところ近づくだけで泳ぎを止めてくれるようになったという。研究者は「彼らが私たちをデカい掃除魚だと思っているのでしょう」と話す

180
8,147
66,719
2,585,657
Ph.D. dolphin retweeted

Jun 12
1
1
591
東大、東京科学大レベルの一般枠でボーダーギリギリで入学できる学生がガチ勢じゃないわけないでしょ・・・
まず2次試験受けられる時点でめちゃくちゃ優秀なんですよ
流石に受験生に対して失礼すぎる。
Jun 12
女子枠があるから入れない、とか言ってるボーダーギリギリのはガチ勢ではないでしょ。というかガチでやっててなんでボーダーなのよ
12
138
1,384
137,287
Ph.D. dolphin retweeted

Protein Dynamics Beyond Structure Prediction
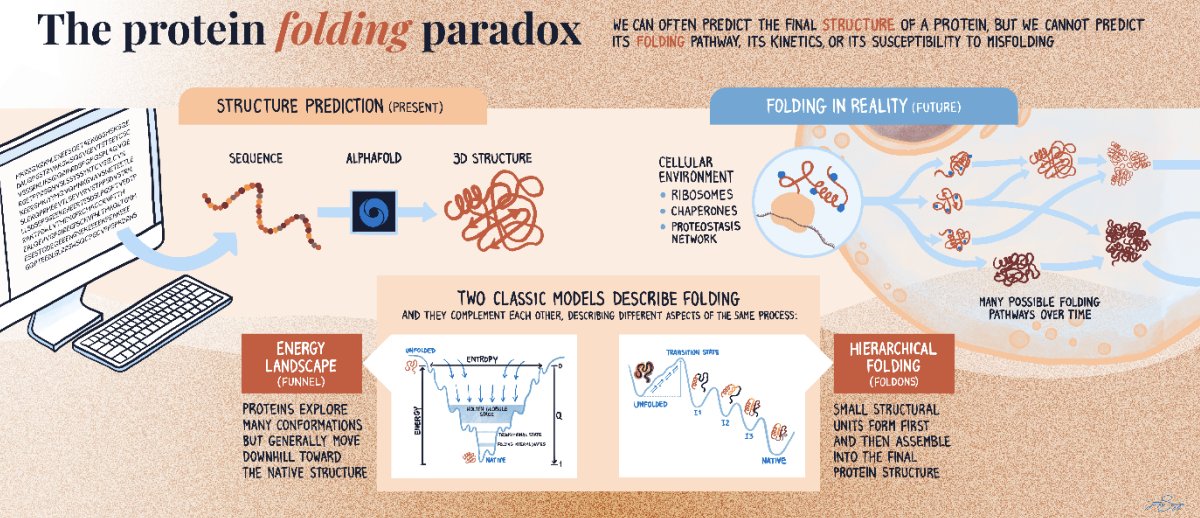
1 The Roadmap argues that AlphaFold-level structure prediction solved “where proteins end up,” but not “how they get there”: folding pathways, kinetics, intermediates, assembly routes, and misfolding competition remain largely non-predictive from sequence alone.
2 Central thesis: proteins are dynamic, stochastic ensembles shaped by context (translation, chaperones, crowding, membranes, PTMs, stress), so a single static structure is an incomplete target; the real target is an evolving conformational landscape plus transition kinetics.
3 It frames a “structure prediction paradox”: models can often predict one (or a few) stable states, yet cannot predict (i) the full ensemble, (ii) barrier heights and rate constants, (iii) pathway heterogeneity, (iv) aggregation propensity, or (v) cellular outcomes of kinetic partitioning.
4 A key emphasis is multiscale coupling: Å-level interactions and ns–µs fluctuations connect to ms–s folding transitions, while misfolding/aggregation can accumulate over years; bridging these time/length scales is positioned as the core methodological gap.
5 The article highlights why in vivo folding is fundamentally different from typical in vitro refolding: co-translational, vectorial emergence from the ribosome; codon-dependent translation speeds; ribosome exit-tunnel constraints; and early engagement by ribosome-associated factors and chaperone systems that reshape pathways rather than merely stabilizing endpoints.
6 Misfolding is treated as a kinetic competition problem, not a “wrong structure” problem: the native fold can be correct yet still lose to off-pathway aggregation under stress/aging or limited proteostasis capacity; amyloid polymorphism (many distinct fibril folds from the same sequence) is presented as a major unpredictability from static models.
7 Translational motivation: for Alzheimer’s, ALS, systemic amyloidoses, CFTR misfolding, and proinsulin variants, detecting aggregated proteins or biomarkers is often insufficient to predict trajectory; the Roadmap argues for mechanistically grounded markers tied to specific conformers and kinetic vulnerabilities.
8 Experimental direction: the field is moving from ensemble averages (CD, FTIR, stopped-flow, NMR, HDX-MS, SAXS/cryo-EM) toward systematic single-molecule trajectory measurements (single-molecule fluorescence/FRET; force spectroscopy such as optical tweezers/AFM) to directly observe intermediates, heterogeneity, and barrier crossings.
9 Computational direction: atomistic MD offers mechanism but is timescale-limited; coarse-grained/multiscale models extend reach but need validation; machine learning is positioned as a bridge to integrate heterogeneous data and accelerate sampling—yet predicting dynamics is described as a fundamentally different challenge than predicting structures.
10 Proposed ecosystem shift: replicate what enabled structure prediction (shared standards, curated datasets, benchmarking) but for folding trajectories and kinetics; the Roadmap advocates iterative experiment–model feedback loops to connect sequence to pathways, ensembles, assembly/misfolding, and ultimately cellular phenotypes.
📜Paper: arxiv.org/abs/2606.08647
#ProteinFolding #ProteinDynamics #SingleMolecule #MolecularDynamics #MachineLearning #Proteostasis #Chaperones #Misfolding #Amyloid #ComputationalBiology
9
32
2,624
この説明も最もですし、更にいえば男女一括で入試してればガチだろうとなかろうと「同じ入試で負けたなら仕方ない」と男子学生は引き下がれますが、「女子枠作って自分より簡単な試験で合格する奴に熱意もなかった」とかマジメに夢に向かって勉強してきた男子高校生があまりにも不憫だと思います
Jun 11

これも話の順番を考えて欲しいんですけど、普通に男女一括で入試をしていたら男女問わずある基準のガチさの人が入ってくるのに対し、特別に女性枠を作ったらガチ度の低い人が入ってくる(ガチ度の高い男子が抜ける分)という話をしているだけで、女性の中でのガチさの違いを言ってるわけじゃないんです。
3
300
Ph.D. dolphin retweeted

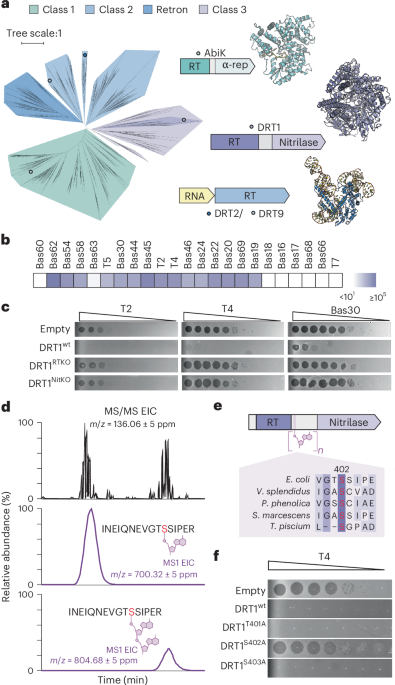
New online! Semirandom DNA adducts regulate a filamentous defense-associated reverse transcriptase dlvr.it/TSywMh
9
32
2,505
Ph.D. dolphin retweeted

Jun 10
【注目プレスリリース】タンパク質液滴の運命はサイズで決まる ―微細加工技術を用いてアミロイド形成を左右する新たな競合過程を発見― / 東北大学,東京科学大学,産業技術総合研究所,神戸大学,大阪大学,筑波大学 research-er.jp/articles/view…
13
38
6,480
まともな女性には是非とも男子高校生の立場になって冷静に考えて欲しいのだが
・貴女の第一志望の大学は男女比率女性8割
・学部の男子学生が「男子俺1人の授業とかあっていづらいんだよね」って言ってる。
これで「来年貴女が合格できる枠を2割減らします」とか大学に言われて納得できるんですか?
13
52
1,511
選択で理科科目選べるのは良くあるけど、大体の学部は化学or物理では?
建築学科に数1Aと生物受験で入学してどうするんでしょう?
Jun 9
設置されているのが生物 / 情報 / 建築とある程度女性人気が高い学科のみで
入試科目見に行ったら数学はIAのみ、理科は1科目でよくてそりゃあ倍率高くて当然だろうという感想。
それと理工学部なのに選択科目が化学と生物のみで、なぜか物理での受験が不可能な模様。
1
1
332
いや「動画10本作ってました!」とかなら「知らねーけど見逃しただけかな?」ってなるけど、1万本以上作ってて思い当たる動画が話題にならないのは流石に嘘くさい・・・
ネガティブ動画を総裁選1500本、衆院選1万本 福岡拠点に作成、IT会社代表【一問一答】 nishinippon.co.jp/item/15012… #西日本新聞
77

Nature research paper: Cold-induced peptide signalling secures pollen resilience and crop yield
go.nature.com/4x5kCKY
3
33
90
17,866
ケンブリッジ大学とDIOSynVax (DVX)社が開発したAIデザインワクチンが第一相臨床試験に合格した記事。
世界中のコロナウイルスのデータをAIに学習させて開発したワクチンで幅広い変異体に有効な可能性がある。
まだ第一相に合格しただけなので今後に注目
medicalxpress.com/news/2026-…
65
左翼のスゴイ所って、日本中の大多数に嫌われる覚悟でこれやってるんじゃなくて、「支持してもらえる」と思ってやってるんだよね
普通はこんな醜い場面は隠し演説シーンとか編集して拡散するじゃん。デモやってる奴が「最近の流行です笑」とか言って自ら拡散してんのよ
我々と根本的な価値観が違う
Jun 6

これをやった人の異常性って「ネタニヤフ・トランプ」と「高市さん」を並べることなんだよ。
本当にこの異常性が彼ら左派リベラルの間では「常識」なんだろう。
どう考えても戦争なんてまったくやってない高市さんが、戦争をおこしたトランプさんやネタニヤフさんと同列だと「信じて疑わない」のだ。
これって日本共産党の田村さんをスターリンやポルポトといっしょに並べるような「異常なカテゴライズ」なのである。たぶんこの人は一生自分のカテゴライズの異常さには気づかないだろう。
そして絶対にプーチンのような「旧東側と反米の指導者は絶対にここにならばない」のである。
これが日本共産党や左派リベラルの「真実」なんだろう。

1
12
47
2,007
デモやってる左翼や自称リベラルの精神年齢ってマジでバイトテロやらかす大学生とかBeRealで顧客情報を拡散する馬鹿な新卒と変わらん。
馬鹿な大学生が政治にハマって高市さん嫌い拗らせて50歳、歳を取ったのが国会デモとかやってる左翼。
2
8
340