
Associate Professor @Penn • Dept Psychiatry Dept Neuroscience Dept Anesthesiology | Pain • Placebo • Poppies • Psychedelics
Joined March 2016
- Tweets 5,913
- Following 2,225
- Followers 3,728
- Likes 21,728
867 Photos and videos








Pinned Tweet
🧠⚡️💊New @Nature publication !
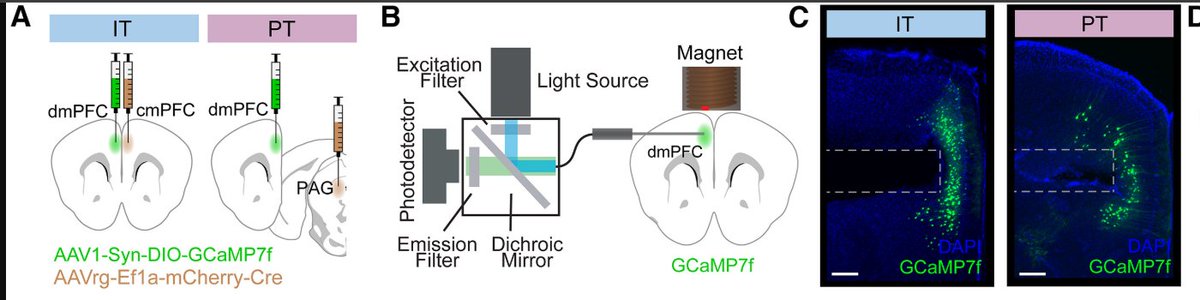
Mimicking opioid analgesia in cortical pain circuits
We built a brain-behavior framework to decode spontaneous chronic pain in mice—and to biologically mimic morphine with a synthetic opioid gene therapy
nature.com/articles/s41586-0…
@PennMedicine

Researchers identify the neurons involved in the emotional distress associated with pain
go.nature.com/3NkL1SB
11
18
89
12,318

Jun 10
saw @A24 Backrooms in this procedurally generated, liminal theater in Pittsburgh .. unsafe vibes

5
305

⚡️GREG_CORDER⚡️ retweeted

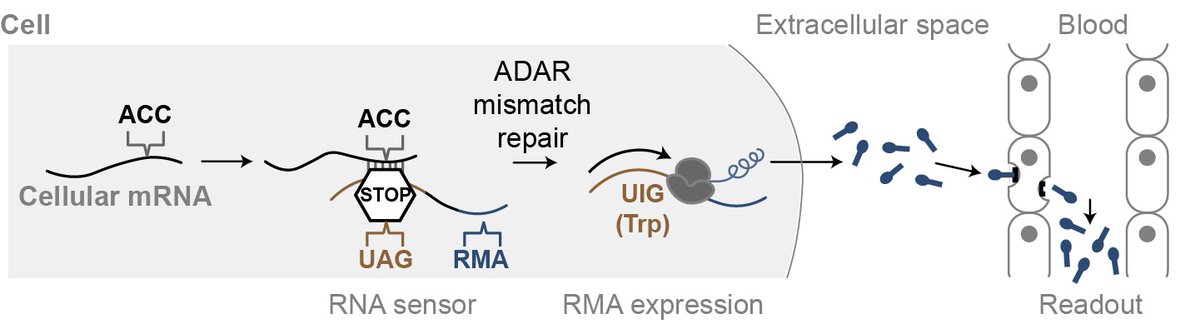
qPCR allowed us to measure transcripts, but just once, destructively, and only in post-mortem tissues. Here, we show we can record transcript level history in vivo and recover this information with a blood test to make a "noninvasive qPCR". nature.com/articles/s41467-0…
3
38
149
13,791
⚡️GREG_CORDER⚡️ retweeted

Our new paper is out in Neuron!
cell.com/neuron/fulltext/S08…
How does the brain decide how much of the past to use when making decisions? In rapidly changing environments, recent experiences matter more; in stable environments, longer histories are useful.
2
49
234
12,340
May 13
Mullet time 🤘🏽🍺

6
748
⚡️GREG_CORDER⚡️ retweeted

May 11
Super excited to share our new preprint!
medrxiv.org/content/10.64898…
We built global reference curves of pain from 6.1M people in 118 countries 🌍 to find out how pain unfolds across the lifespan, and how individual- and country-level risk factors shape these trajectories.
1
14
35
2,990
⚡️GREG_CORDER⚡️ retweeted

May 6
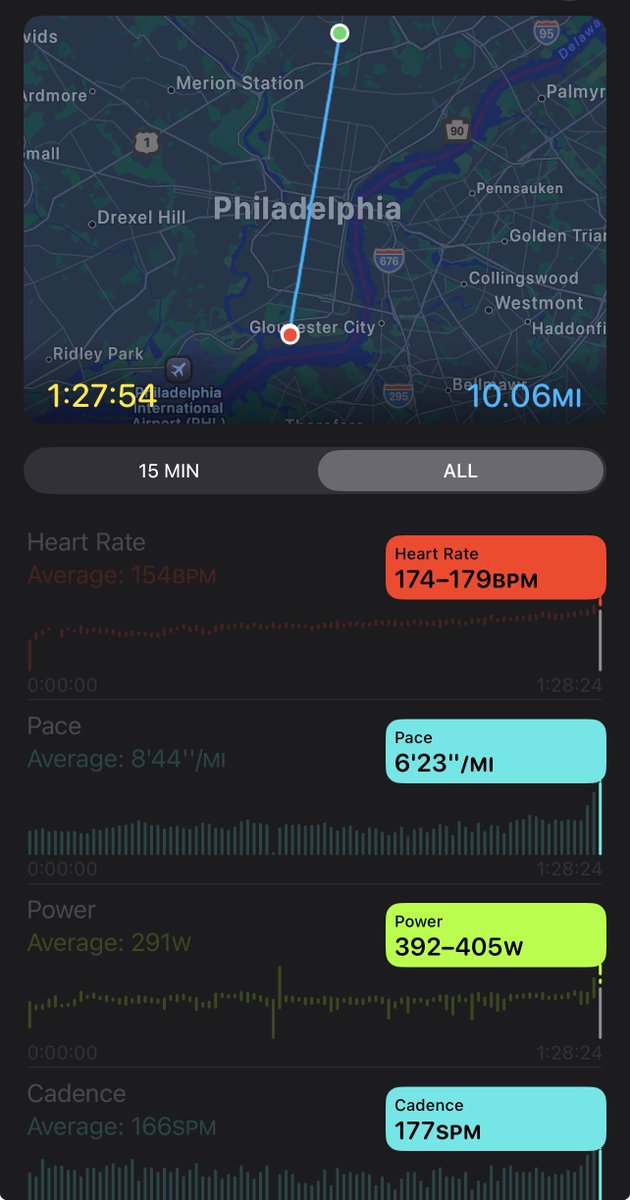
13:28 per mile pace for TWO HUNDRED AND FIFTY MILES 🤯🤯🤯🤯🤯🤯🤯🤯

🏆 Congratulations to Rachel Entrekin for finishing first in this year’s Cocodona 250 and setting a new overall course record of 56:09:48!
The Cocodona course stretches roughly 250 miles from Black Canyon City to Flagstaff. Unlike traditional ultramarathons, Cocodona takes place over the course of several days, turning the race into a battle of endurance, logistics and mental resilience.
18
27
1,316
217,631
7 weeks post meniscus injury .. I’ll take it 🙏🏼
Broad Street 10 Miler 🦅


3
20
1,777
⚡️GREG_CORDER⚡️ retweeted

Apr 26
OMG.
🇰🇪 Sabastian Sawe becomes the first man ever to break 2 hours in a marathon (legal conditions) in 1:59:30 at the London Marathon!
Yomif Kejelcha 🇪🇹 runs 1:59:41 in his DEBUT.
Jacob Kiplimo 🇺🇬 takes third in 2:00:28
All under the previous WR.
265
2,703
24,564
3,896,710
Apr 25
check out this piece for @SfNtweets on my grad student @neuro_melody ❤️
sfn.org/publications/neurosc…
1
4
15
1,334
⚡️GREG_CORDER⚡️ retweeted

Mar 10
Pleased to share our latest work by Lily He, Parth Bhatia, Shams Bhuiyan et al. combining human and mouse DRG multi-omics with in vivo AAV screening to identify enhancers that bias gene expression toward distinct classes of nociceptors. @NIH_NINDS & HEAL
biorxiv.org/content/10.64898…
2
11
31
2,543
Apr 24
Really beautiful rigorous work from the Liston and Levitz Labs combining circuit, molecular, and behavioral approaches to dissect the role of μ-opioid receptors (MOR/OPRM1) in ketamine’s antidepressant effects
One of the central findings is that MORs are enriched in somatostatin (SST) interneurons in cortex, and that these cells play a key role in mediating ketamine’s behavioral effects
There is a lot to like here: the study is technically sophisticated, the circuit logic is compelling, and the SST-dependent mechanism is supported by multiple independent experiments
But stepping back — it’s also critical that we get the cell-type distribution of OPRM1 correct, because this has direct implications for how we think about:
• opioid analgesia
• opioid use disorder
• cortical circuit modulation
• and the design of next-generation MOR-targeting therapeutics
And on that specific point, I think there is a non-trivial dataset interpretation issue worth discussing
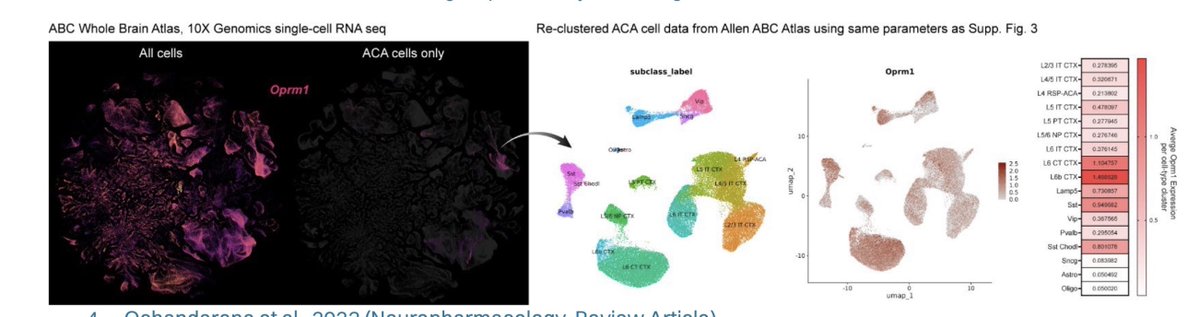
The key claim relies heavily on older SMART-seq datasets (ACA/ALM; ~5K cells) from the Allen Institute
These datasets were incredibly important at the time — but they were not designed as unbiased quantitative cellular censuses
A few technical considerations that matter a lot for a gene like Oprm1:
1) Sampling design (not a census)
These datasets rely on:
• targeted dissections (ACA/ALM only)
• FACS-based sorting
• heavy use of transgenic driver lines
This means the data reflect what was selected, not necessarily the true population distribution
2) Cell numbers and statistical power
~5,000 ACA cells total. Once subdivided across excitatory subclasses, power drops quickly
For low-abundance GPCR transcripts like Oprm1, this creates:
• dropout sensitivity
• threshold artifacts
• unstable “presence/absence” calls
• inflated apparent enrichment in small populations
3) SMART-seq recovery biases
Requires intact dissociated cells:
• large projection neurons underrepresented
• pyramidal neurons more fragile
• interneurons often more recoverable
4) Quantification annotation (2018-era pipelines)
Older gene models isoform collapsing exon/intron handling can all affect detection of isoform-complex GPCRs like Oprm1
Now contrast that with newer datasets:
The Allen Brain Cell (ABC) Atlas includes ~4 million cells across the entire mouse brain, with:
• orders-of-magnitude larger sampling
• stable estimates across cortical subclasses
• improved taxonomy
• spatial integration
And importantly — across this dataset, OPRM1 signal is not restricted to SST interneurons
We see the same pattern across:
• our own single-nucleus RNA-seq (100k cells)
• Allen ABC Atlas (4 million cells)
• multiplexed FISH
• immunohistochemistry
• n=4 Oprm1-Cre mouse lines
• MORp viral promoter strategies
Across all of these orthogonal approaches, the result is highly consistent:
→ OPRM1/MOR is enriched in glutamatergic cortical populations, not exclusively confined to SST interneurons
So how do we reconcile this?
The most parsimonious explanation is not that either dataset is “wrong,” but that they are answering different questions under different constraints:
SMART-seq (2018):
→ high depth, small N, targeted sampling
→ vulnerable for sparse genes
ABC-scale (modern):
→ massive N, robust population estimates
→ better suited for cell-type distribution
For genes like Oprm1 — low abundance, heterogeneous, biologically critical —
scale sampling design cross-modal validation are decisive
The broader point:
As a field, we need to be careful about making strong claims about cell-type specificity of neuromodulatory receptors based on early-generation datasets — especially when:
• atlas-scale data now exist
• multiple orthogonal methods converge
• and the implications extend to therapeutics
None of this detracts from the importance of SST interneurons in MOR-dependent circuit function — that biology may be very real and important
But the global cortical distribution of OPRM1 appears broader, and heavily includes glutamatergic neurons
And that distinction matters for new appraoches for depression, pain and OUD
Apr 23

Mechanism-guided identification of antidepressant G protein-coupled receptor drug targets: Cell cell.com/cell/fulltext/S0092…
4
32
5,292
⚡️GREG_CORDER⚡️ retweeted

Apr 20
"When scientists are absent from public conversations, misinformation fills the space". We have to discuss science openly in public. That means not just advertising & hyping & retweeting but also educating, discussing, criticizing, defending, arguing. All of it.
Apr 18

The hill I will die on - we have to rethink graduate training.
“Scientists are trained for a world where data speaks for itself. Where misinformation moves slowly. Where scientific expertise naturally rises above noise. That world is gone.”
sciencepolitics.org/2026/03/…

3
10
56
4,811
Apr 17
🔥🍾🙌
Huge congrats to Dr. Sophie Rogers (@synaptic_soph) receiving the Saul Winegrad Award for Outstanding Dissertation !!!
🧠@Penn @pennbgs @PennNGG
A truly field-defining body of work on pain, psychedelics, and cortical computation (first authorships in Nature and Nature Neuroscience)
Couldn’t be a more proud mentor 🥹
Check out Sophie's thesis work here:
- nature.com/articles/s41586-0…
- nature.com/articles/s41593-0…
6
59
3,933
Apr 16
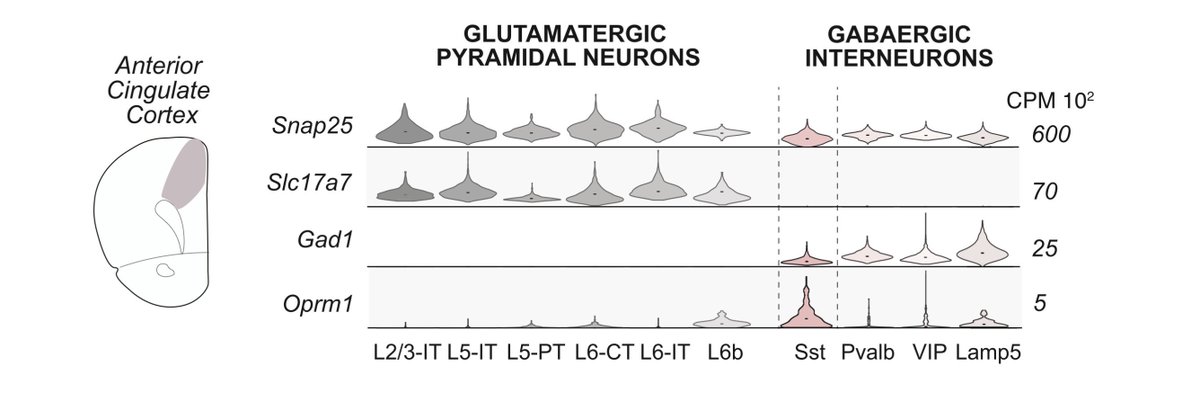
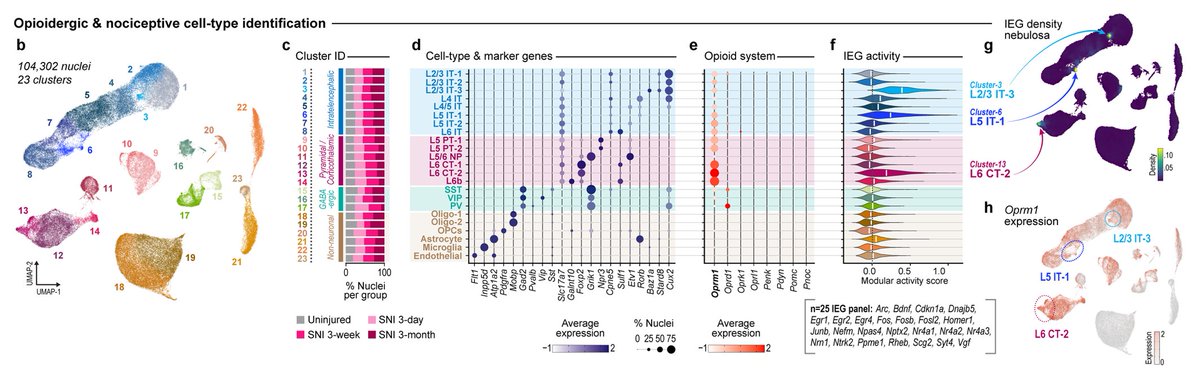
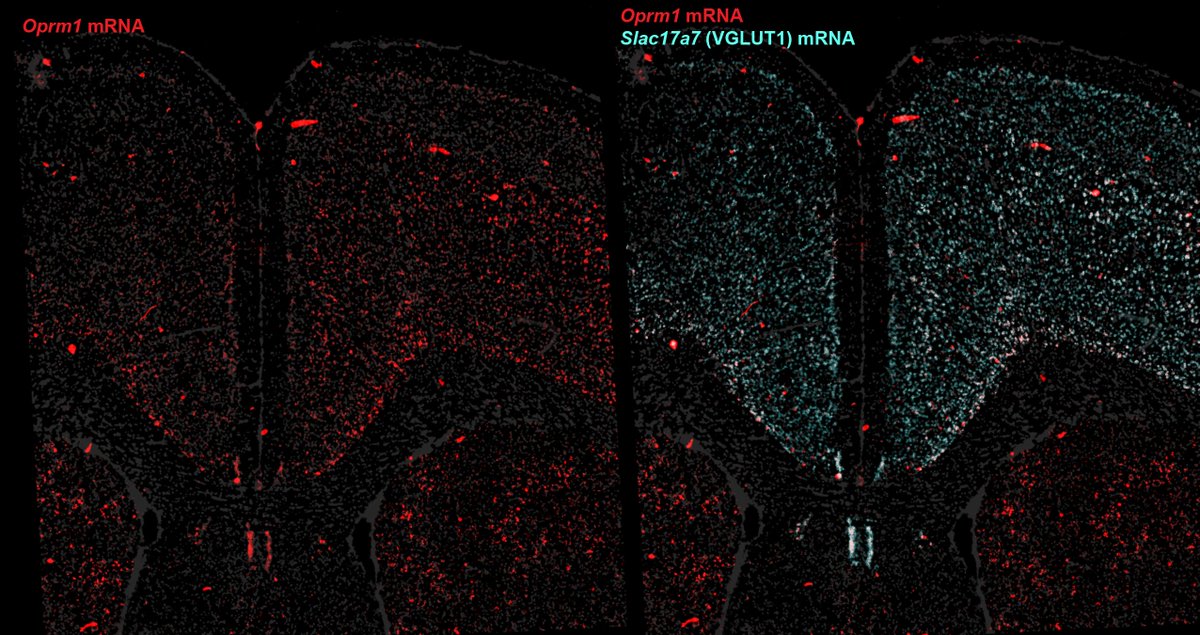
Across single-nuc seq, FISH, IHC, four cell-type mouse lines, and a viral promoter, OPRM1/MOR is consistently enriched in glutamatergic PFC neurons
While some slice electrophysiology studies emphasize GABAergic localization—biased, low throughput methodological differences?

5
3
37
7,244
Apr 17
we need multiple, orthogonal approaches 👇
x.com/FlyBottleEscape/status…
Apr 17
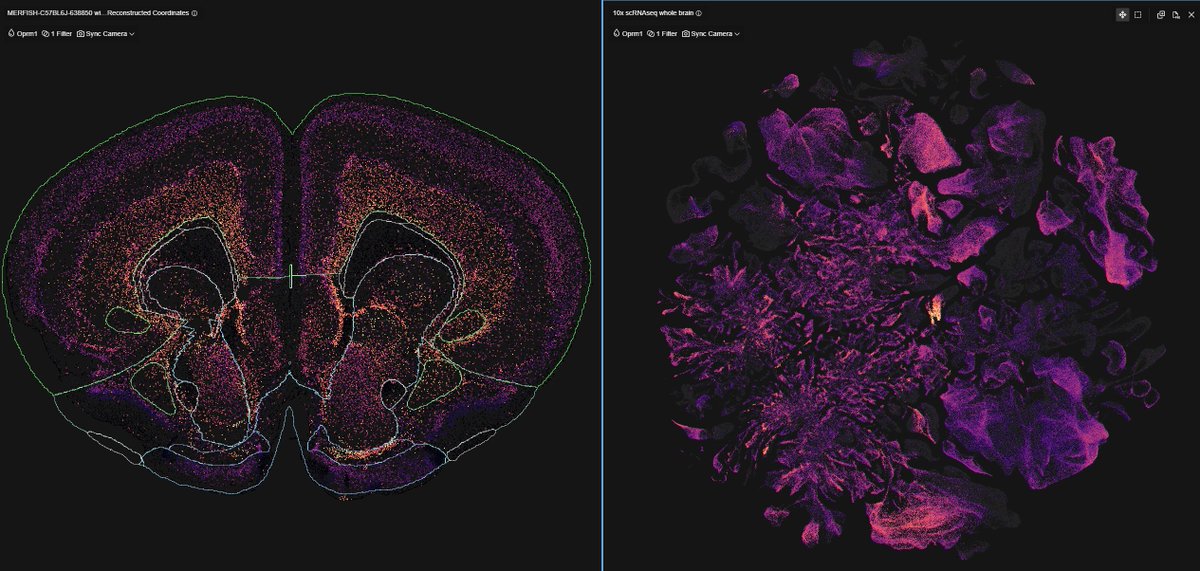
1. That is not a 'single nuc' dataset ... What’s being shown here is from the 2006 Allen Mouse Atlas using chromogenic DIG riboprobe ISH(single probe, single section, enzyme-amplified signal) ... That’s fundamentally different from modern datasets (snRNA-seq, MERFISH, Xenium), which sample 10⁵–10⁶ cells across animals with cell-type classification.
That legacy ISH pipeline has known limitations for genes like GPCRs:
- single probe per gene (isoform / region sensitivity)
- nonlinear enzymatic amplification (BCIP/NBT)
- limited dynamic range and no cell-type annotation
- section-level variability (one slice ≠ population estimate)
So a single probe image from 20 years ago is not a rigorous argument nor a quantitative or cell-type–resolved readout
2. Re: “can snRNA-seq detect GPCRs/neuropeptides?”
Yes—with caveats. Nuclear RNA has lower abundance, but
- detection improves with depth and aggregation across cells
- multiple platforms (snRNA-seq, MERFISH, Xenium) now converge on similar cell-type assignments
- MERFISH/Xenium bypass dropout via targeted probe amplification and spatial counting
So the question isn’t one dataset—it’s convergence across modalities.
3. For Oprm1, we see consistent enrichment in glutamatergic populations across
- snRNA-seq (mouse human)
- spatial transcriptomics (MERFISH / Xenium)
- IHC
- genetic access (multiple Oprm1-Cre lines)
- viral promoter strategies
- in vivo calcium imaging (morphine modulation of mMORp pyramidal activity)
That’s orthogonal convergence, which is not cited ever in your papers/review artciles
4. Electrophysiology is powerful, but it’s also sampling-limited
- biased toward visually accessible neurons (often L2/3 but not layers 5-6, as written in your Methods)
- low N relative to transcriptomic datasets (10s vs 100k-millions of cells)
- cell identity often inferred post hoc
So slice ephys can overrepresentspecific populations depending on targeting strategy
MOR signaling in cortex is largely dendritic and presynaptic, not confined to somatic excitability. Acute slices sever long-range afferents and axonal projections, limiting access to these compartments.
Patch-clamp readouts (spiking, IPSCs) capture only a subset of GPCR function, missing modulation of dendritic integration, Ca²⁺ dynamics, and release probability, thus requiring orthogonal readouts to make claims like where MORs are expressed .. luckily that data exists! and it shows OPRM1/MORs are in gluatmatergic neurons and to a lesser extent some interneurons populations
1
1,193
Apr 17
1. That is not a 'single nuc' dataset ... What’s being shown here is from the 2006 Allen Mouse Atlas using chromogenic DIG riboprobe ISH(single probe, single section, enzyme-amplified signal) ... That’s fundamentally different from modern datasets (snRNA-seq, MERFISH, Xenium), which sample 10⁵–10⁶ cells across animals with cell-type classification.
That legacy ISH pipeline has known limitations for genes like GPCRs:
- single probe per gene (isoform / region sensitivity)
- nonlinear enzymatic amplification (BCIP/NBT)
- limited dynamic range and no cell-type annotation
- section-level variability (one slice ≠ population estimate)
So a single probe image from 20 years ago is not a rigorous argument nor a quantitative or cell-type–resolved readout
2. Re: “can snRNA-seq detect GPCRs/neuropeptides?”
Yes—with caveats. Nuclear RNA has lower abundance, but
- detection improves with depth and aggregation across cells
- multiple platforms (snRNA-seq, MERFISH, Xenium) now converge on similar cell-type assignments
- MERFISH/Xenium bypass dropout via targeted probe amplification and spatial counting
So the question isn’t one dataset—it’s convergence across modalities.
3. For Oprm1, we see consistent enrichment in glutamatergic populations across
- snRNA-seq (mouse human)
- spatial transcriptomics (MERFISH / Xenium)
- IHC
- genetic access (multiple Oprm1-Cre lines)
- viral promoter strategies
- in vivo calcium imaging (morphine modulation of mMORp pyramidal activity)
That’s orthogonal convergence, which is not cited ever in your papers/review artciles
4. Electrophysiology is powerful, but it’s also sampling-limited
- biased toward visually accessible neurons (often L2/3 but not layers 5-6, as written in your Methods)
- low N relative to transcriptomic datasets (10s vs 100k-millions of cells)
- cell identity often inferred post hoc
So slice ephys can overrepresentspecific populations depending on targeting strategy
MOR signaling in cortex is largely dendritic and presynaptic, not confined to somatic excitability. Acute slices sever long-range afferents and axonal projections, limiting access to these compartments.
Patch-clamp readouts (spiking, IPSCs) capture only a subset of GPCR function, missing modulation of dendritic integration, Ca²⁺ dynamics, and release probability, thus requiring orthogonal readouts to make claims like where MORs are expressed .. luckily that data exists! and it shows OPRM1/MORs are in gluatmatergic neurons and to a lesser extent some interneurons populations

Since you are attacking the rigor of our work I'll return a question
Why isn't Oprl1 showing up in this single nuc dataset? Oprl1/NOPR is abundant in PFC mouse.brain-map.org/experime…
Does snRNA-seq have the sensitivity needed to accurately measure GPCRs and neuropeptides?
3
14
12,869
Apr 17
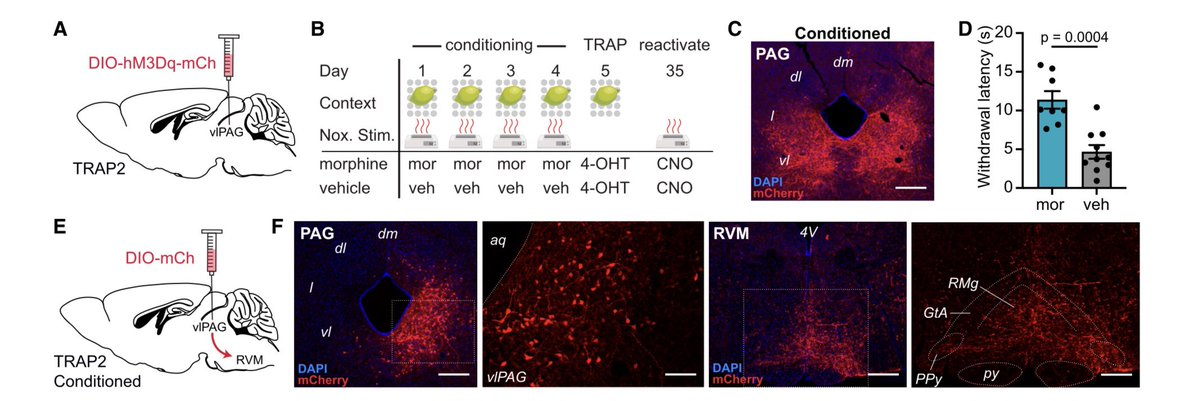
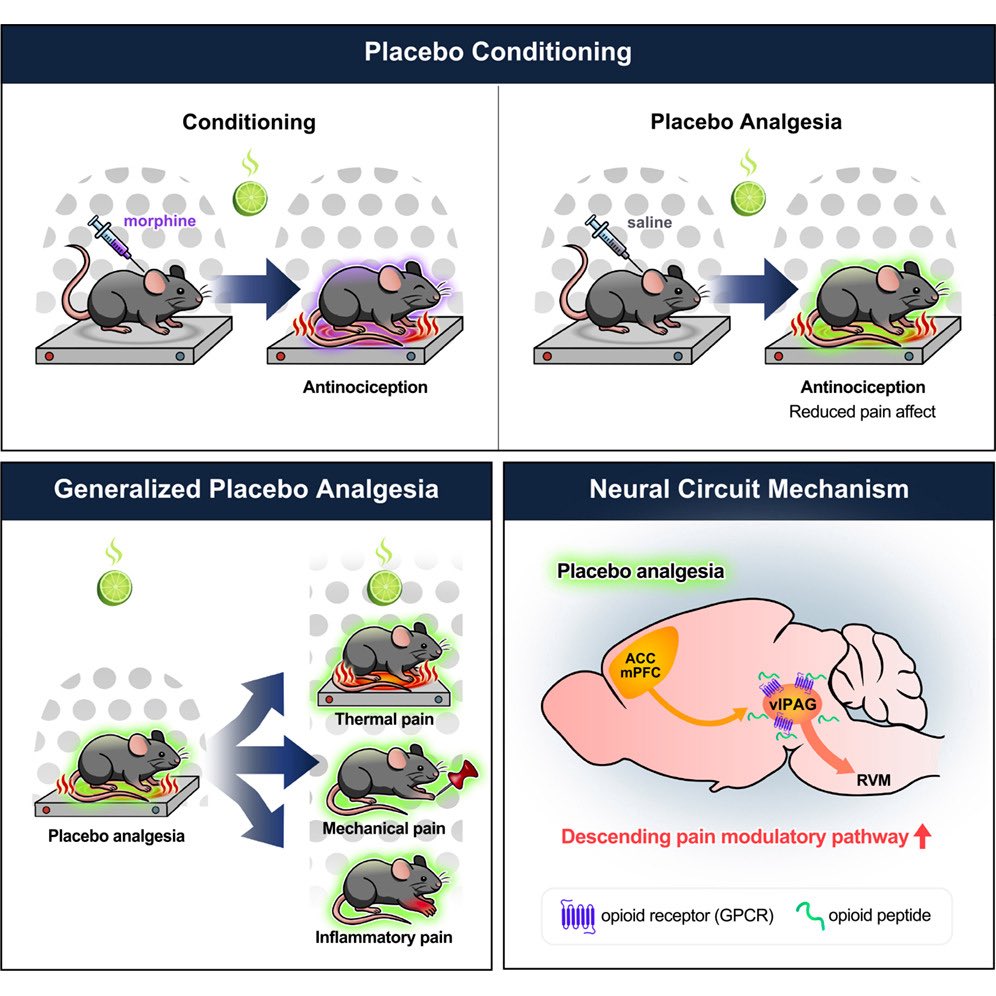
Check out the new @chromism placebo paper in @NeuroCellPress ! 🔥
Stoked that our lab could contribute 👇🏼
Activity capture chemogenetic reactivation of a placebo analgesia ensemble in the vlPAG drives on-demand pain relief
💊 🧠 ⚡️
cell.com/neuron/fulltext/S08…
@PennMedicine

1
11
54
4,079
⚡️GREG_CORDER⚡️ retweeted

Now that it has been out for a week, I thought I’d address a few strange takes regarding our recent mega-analysis on the effects of psychedelics on brain function (specifically, resting-state functional connectivity). 1/15
nature.com/articles/s41591-0…
3
10
72
10,352