
Building AI for biological design @biohub (fka @evoscaleai) 🧬 Prev: @Theteamatx 🌘 PhD @UCBerkeley 🙈
Joined November 2015
- Tweets 461
- Following 1,448
- Followers 1,038
- Likes 1,422
55 Photos and videos






Pinned Tweet

May 27
It's been exhilarating to watch this model get better and better, and I’m grateful to work with such an incredible, cross-disciplinary team across folding, binder design, and interpretability! This paper also sets a new scaling law for papers, compressing 3 papers into 1.
May 27

Today we're announcing ESMFold2, an open scientific engine to power prediction, design, and discovery across protein biology.
The new model delivers state of the art performance on protein interactions, especially antibodies, a critical modality for therapeutics.
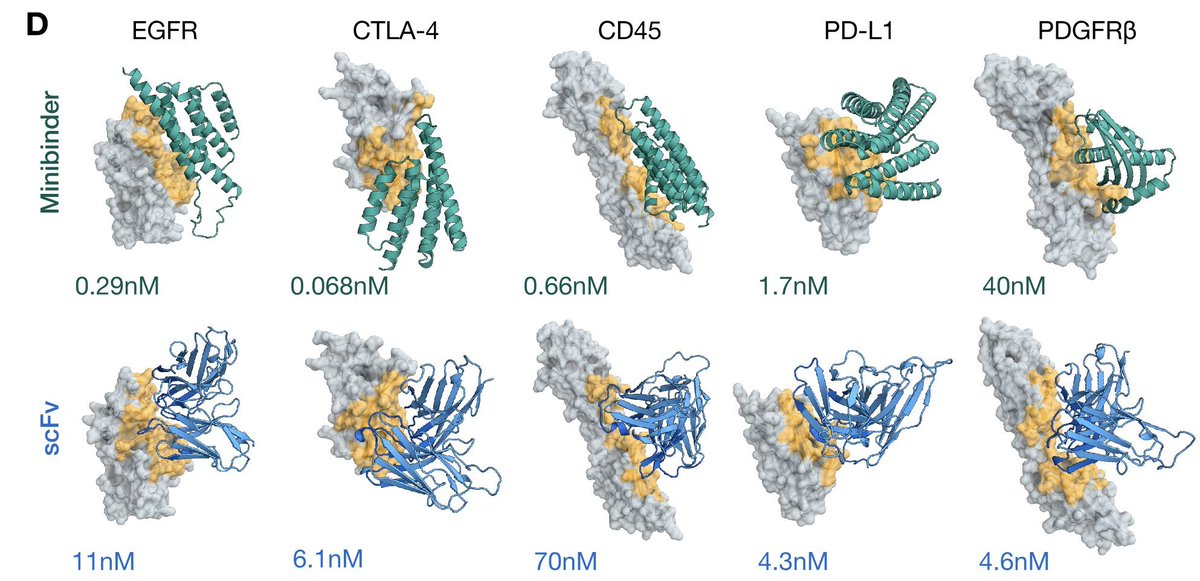
We have designed and validated miniprotein binders and single chain antibodies across five therapeutic targets that are important in cancer and immunology. We are seeing very high success rates, and affinities at levels consistent with therapeutic activity.
We’re also releasing an atlas of 6.8 billion proteins, and 1.1 billion predicted structures.
ESMFold2 is built on a state of the art language model that has been trained on billions of protein sequences.
A world model of protein biology emerges through language modeling.
We’ve used the techniques of mechanistic interpretability developed to understand large language models to understand the concepts ESM uses to represent proteins.
The model’s representation space has a compositional organization of features across scales, levels of complexity, and abstraction, that reflects and mirrors the understanding of protein biology developed through a century of empirical science.
This understanding emerges without prior knowledge, just from language modeling of protein sequences.
Language models are becoming a powerful substrate to understand and program biology.
The design of protein interactions is one of the most fundamental problems in biophysics, and has critical implications for the discovery of new medicines. A simple gradient based search with the model was able to discover high-affinity protein binders.
I'm excited by the potential this has to accelerate basic science and the understanding of proteins. And especially for the new avenues it opens up for therapeutic design and medicine.
6
4
65
5,161
Jun 11
Imagine being able to see every protein in a cell at 3Å resolution.
Excited for these datasets to come online! Just don't look at the laser...
Jun 11
Together with UC Berkeley we are announcing the laser phase plate - a breakthrough in atomic resolution imaging. This is the brightest continuous wave laser in the world, 100 million times the intensity of the surface of the sun.
Phase contrast plays an important role in microscopy, but it was thought close to impossible for electron microscopy, where it would require interfering with an electron beam. Holger Mueller and Robert Glaeser proposed exactly this using a standing wave laser. It has taken over 15 years to make this a reality. Biohub partnered with UC Berkeley and Mueller to support this work and to engineer and build the technology.
Contrast has been the critical barrier to achieving atomic resolution imaging of the cell. In cryo-electron tomography, a cellular imaging technology that uses electron microscopy, the low contrast makes it impossible to resolve anything but the largest proteins within their cellular context. The laser phase plate removes that barrier.
With advances in AI this breakthrough in contrast will start to open up a new frontier in structural biology, that will allow us to see the molecular machines of the cell, and how they assemble into far more complex and dynamic systems, and understand how they work.
5
108
7,784
Neil Thomas retweeted

Jun 8
Antibody LMs learn what looks antibody-like, but not how selection turns naive germline antibodies into strong binders.
@aakarshv1 and I are excited to share CoSiNE, a model that learns this germline-to-mature process for variant effect prediction and antibody design. (1/8)
8
50
221
41,512
Neil Thomas retweeted

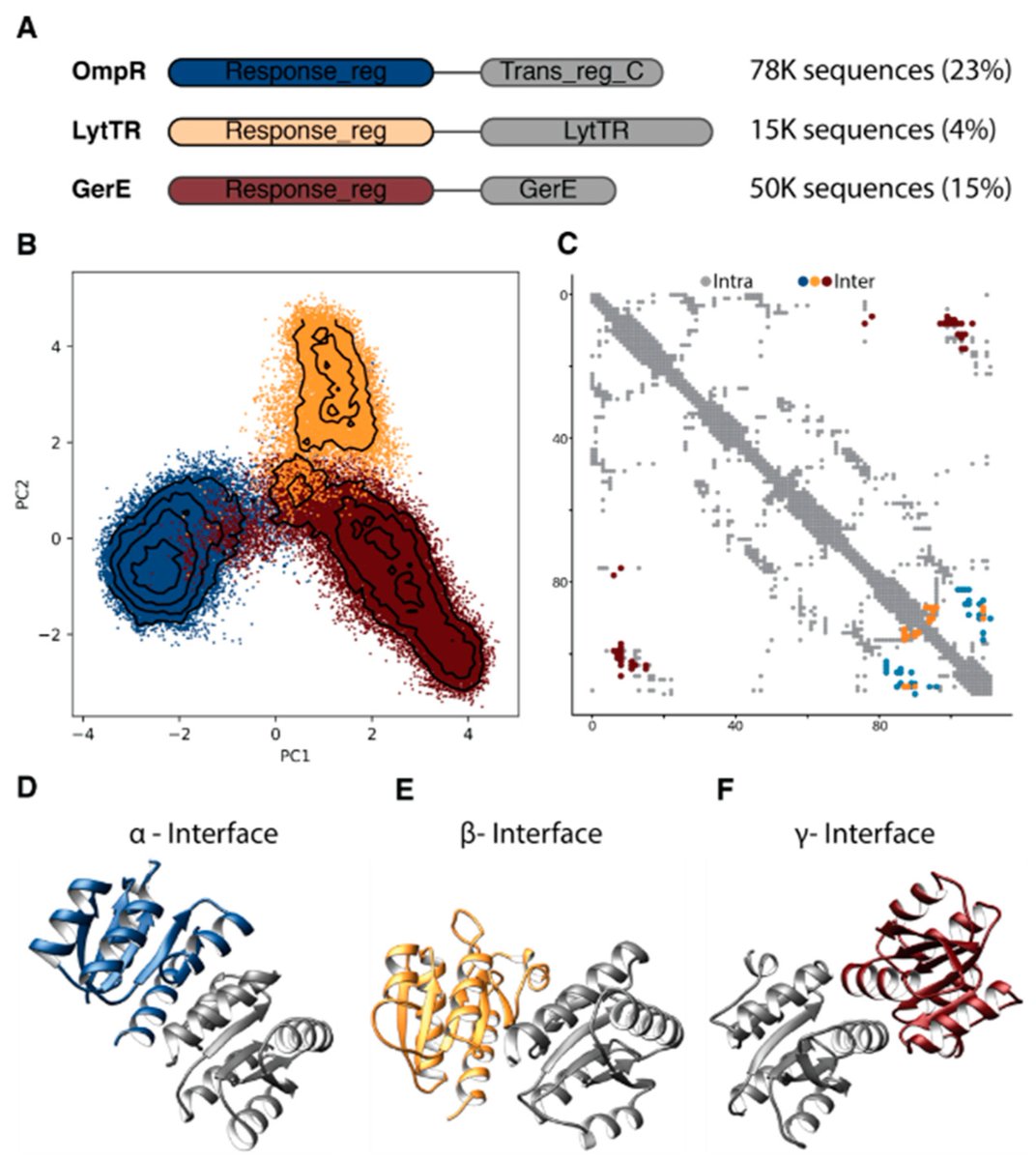
Screen 1M random protein sequences to discover that biology-like folds are accessible from random sequences with surprising frequency
@KlaraH_lab

3
57
319
29,250
Jun 8
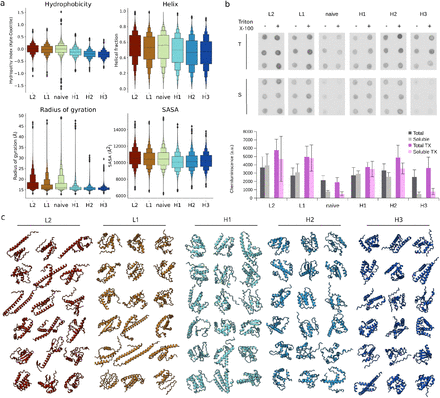
Max-pooling sparse features > mean-pooling dense features
Pooling nerds will enjoy this part of the ESMC interpretability work.
Protein language models have to capture *everything* about a sequence, including the organism that the sequence came from, its structural fold, etc in order to be able predict amino acids.
If a functional signal (e.g. active site, allostery) is local in sequence space, it may be drowned out by mean-pooling.
Max-pooling across sparse features preserves this signal, and we show that it improves functional homolog recovery in the presence of decoys.
Jun 5

Furthermore, if we maxpool the features across the sequence dimension, we reduce the effect from low frequency averaging of dense embeddings, and get a vocabulary of strongly activated functional features for each protein.
In fact, now we can use some of the common techniques found in information retrieval, such as TF-IDF and BM25 [our Jaccard similarity is close to this] for protein search.
2
10
66
4,131
Jun 8
I think there is a nice theoretical relationship between the intuition above and the "APC correction" used to remove spurious phylogenetic signal from contact maps, but I will let someone smarter than myself figure it out.
x.com/ebetica/status/2062943…
Jun 5
Random aside: Qin and Cowell (2019) was a paper that @sokrypton showed me which described that the MSA correlated with contacts only after removing first principal components. The low frequencies signals learned by LLMs tend to be around phylogeny, followed by contact, whereas function is incredibly high frequency.

1
510
Neil Thomas retweeted

Jun 5
🧵 around the interpretability work that helps connect ESMC embeddings to natural language - protein function at the micro level is around residue level mutations but at the macro level is around how they behave in the real world.

One early finding: evolutionary links between gene-editing enzymes across completely different branches of life — connections nobody had made before. This is what becomes possible when you can question protein space at scale, not just search it.
Explore ESM Atlas: bit.ly/4dJcF6G
3
15
71
7,510
Neil Thomas retweeted
Jun 2
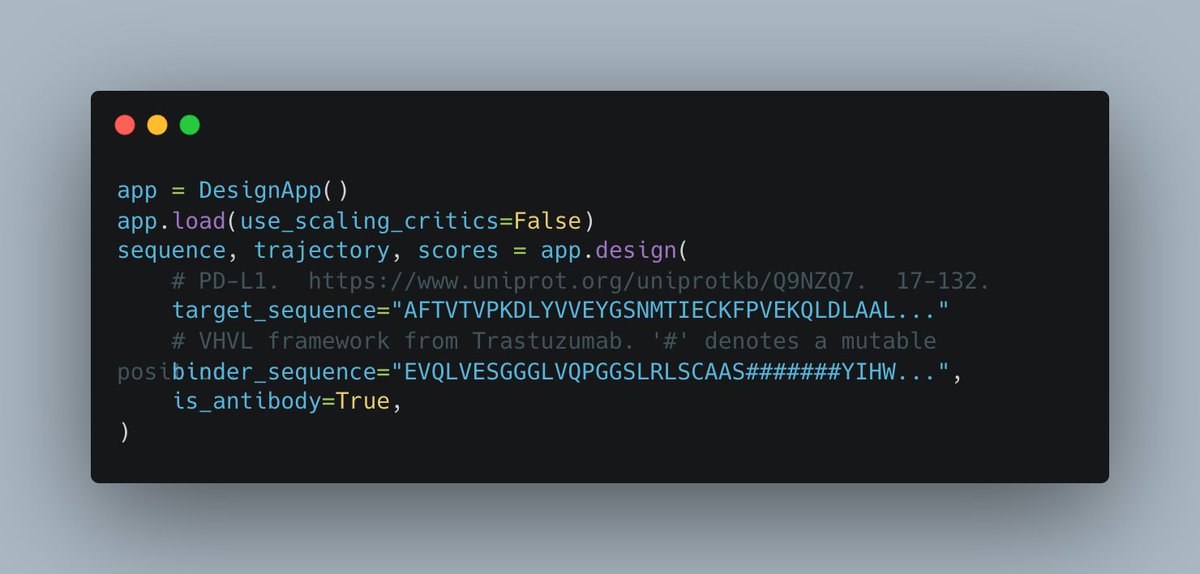
How to design your own PD-1 binder in 4 easy steps:
1. Download the tutorial notebook from the ESM team
2. Get a @modal API key to scale it up
3. Scaling it up, O($1000) will get you a 96 well plate of minibinders with >50% success rates on typical targets
4. Test it in the lab!
5
15
91
10,161
Neil Thomas retweeted

Jun 3
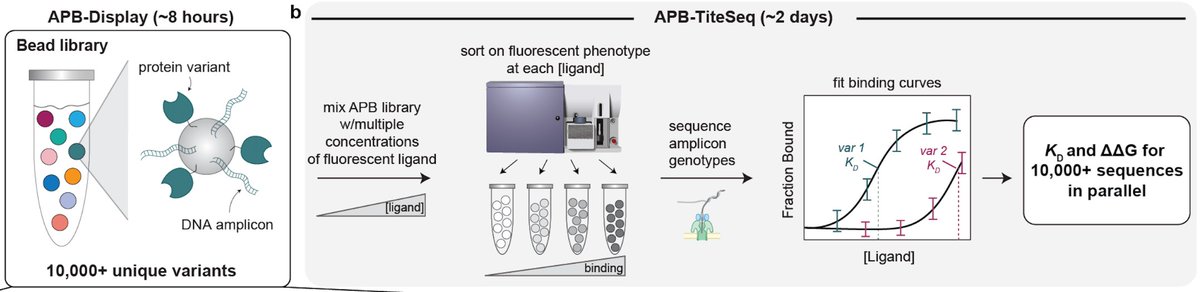
Characterizing AI-designed proteins requires quantitative biochemistry at massive scale. Enter Amplicon/Protein Bead Display (APB-Display), a fully in vitro platform that quantifies Kd's for >100,000 variants in <3 days (preprint link below!) @Stanford_ChEMH @czbiohub (1/n)
3
97
439
62,329
Jun 3
ESMFold2 can be inverted to design new protein binders including miniproteins and scFvs! Take our protocol for a spin on @modal!
github.com/Biohub/esm/blob/m…
Jun 2

I’m so excited about the launch of ESMFold2, ESMC, and the new ESM Atlas. This was a massive team effort, and I’m grateful to have worked with such an incredible group @biohub.
A headline result I’m especially excited about: ESMFold2 can design minibinders and antibodies with nanomolar affinity, target selectivity, and functional activity against therapeutically relevant targets.
Today, we’re sharing the full binder design protocol.
2
5
68
4,367
Neil Thomas retweeted

Jun 2
I’m so excited about the launch of ESMFold2, ESMC, and the new ESM Atlas. This was a massive team effort, and I’m grateful to have worked with such an incredible group @biohub.
A headline result I’m especially excited about: ESMFold2 can design minibinders and antibodies with nanomolar affinity, target selectivity, and functional activity against therapeutically relevant targets.
Today, we’re sharing the full binder design protocol.
May 27
Today we're announcing ESMFold2, an open scientific engine to power prediction, design, and discovery across protein biology.
The new model delivers state of the art performance on protein interactions, especially antibodies, a critical modality for therapeutics.
We have designed and validated miniprotein binders and single chain antibodies across five therapeutic targets that are important in cancer and immunology. We are seeing very high success rates, and affinities at levels consistent with therapeutic activity.
We’re also releasing an atlas of 6.8 billion proteins, and 1.1 billion predicted structures.
ESMFold2 is built on a state of the art language model that has been trained on billions of protein sequences.
A world model of protein biology emerges through language modeling.
We’ve used the techniques of mechanistic interpretability developed to understand large language models to understand the concepts ESM uses to represent proteins.
The model’s representation space has a compositional organization of features across scales, levels of complexity, and abstraction, that reflects and mirrors the understanding of protein biology developed through a century of empirical science.
This understanding emerges without prior knowledge, just from language modeling of protein sequences.
Language models are becoming a powerful substrate to understand and program biology.
The design of protein interactions is one of the most fundamental problems in biophysics, and has critical implications for the discovery of new medicines. A simple gradient based search with the model was able to discover high-affinity protein binders.
I'm excited by the potential this has to accelerate basic science and the understanding of proteins. And especially for the new avenues it opens up for therapeutic design and medicine.
3
22
82
147,272
Neil Thomas retweeted

May 31
A few edible plants have proteins that sit close to miraculin in the ESM Protein Atlas, so I thought I'd try extracting what protein I could from said plants and tasting it... Anyway, null result but an excuse to muck about :) Video lab notes: youtube.com/watch?v=mwGZb8zw…

5
11
77
11,686
May 30
claude just told me that my proposed algorithm was "numerically dead on arrival"
hope your day is going better
22
10
286
329,425
Neil Thomas retweeted

May 28
I added ESMFold 2 to github.com/hgbrian/foldism -- and some other niceties like an optional reference pdb

1
15
85
3,409
Neil Thomas retweeted

May 29
We built a joint experimental and computational platform for scalable multi-modal single-cell chemical screens — profiling RNA, protein (including phospho-signaling), and chromatin accessibility responses to thousands of small molecule perturbations in parallel. biorxiv.org/content/10.64898…
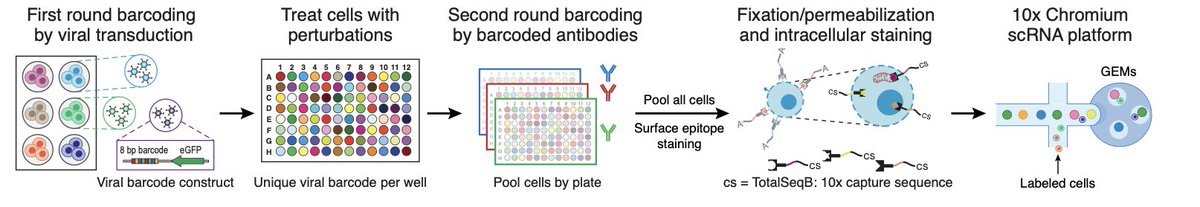
2
40
180
13,657
May 29
.@proteinrosh out here recruiting research partners
May 29

We've done a million of these deep dives into interpreting and understanding ESMC features, it's just that we don't quite know how to write about them other than to say "here are a bunch of cool observations".
1
24
1,461
Neil Thomas retweeted

May 29
We've done a million of these deep dives into interpreting and understanding ESMC features, it's just that we don't quite know how to write about them other than to say "here are a bunch of cool observations".
May 29
Here are all the features that activate on the second arginine in the motif - clearly calling out a relationship to phosphoinositide.
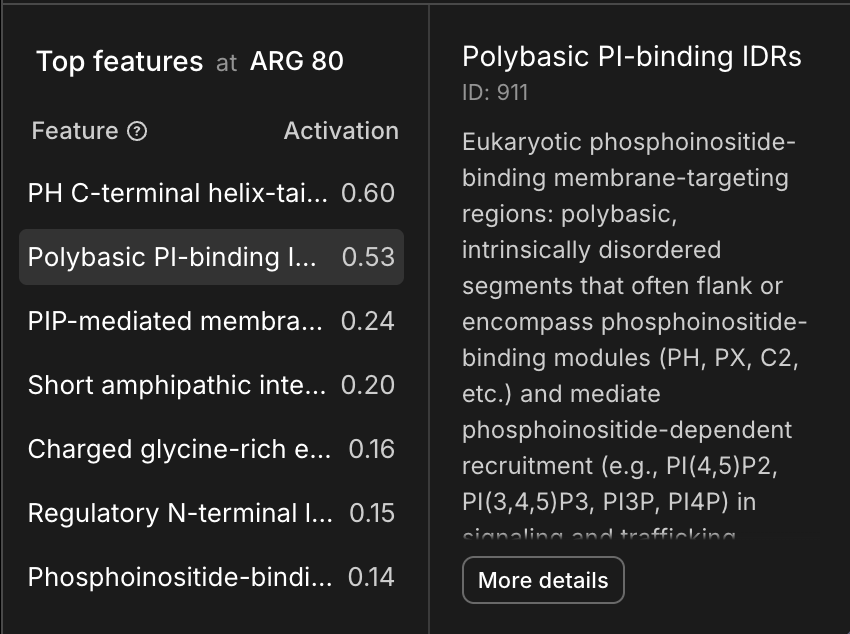
1
2
29
5,915
Neil Thomas retweeted

May 28
Finally got the BDBV Trimer folded properly using esmfold2!
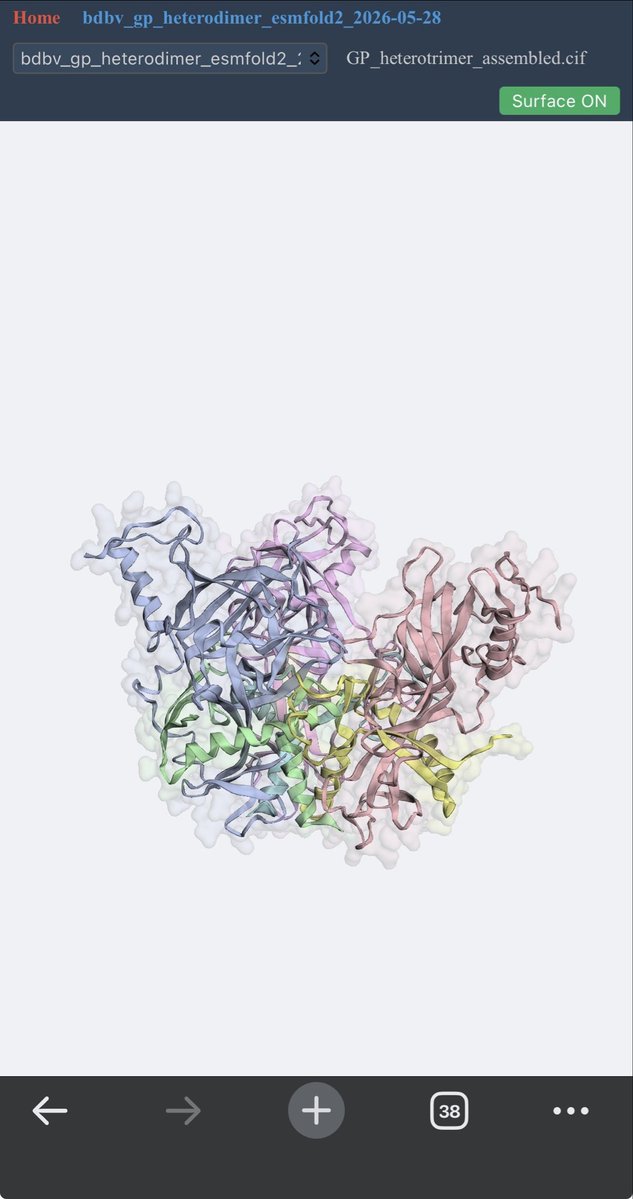
6
35
1,562
Neil Thomas retweeted

May 28
It looks like that made a big difference. It found a much higher confidence pose (0.85 ipTM vs 0.81 with AF3 and 0.8 earlier with ESM2) that actually makes much more sense than the original pose and plausibly explains its MOA. Also no artifacts. Amazing work!
3
14
1,730