
20 Jul 2025
Protein Structure Alignment Significance Is Often Exaggerated
Computational biology researchers have revealed a significant issue in protein structure alignment: the statistical significance of alignments is frequently overestimated. This problem is exacerbated by the vast number of high-quality predicted protein structures from machine learning, where unrelated proteins often show convergent evolution of secondary and tertiary motifs, leading to an excess of high-scoring false positive alignments.
Previous methods for estimating significance, often relying on Gumbel or Gaussian distribution fits, fail to accurately model the high-scoring tail of false positive distributions. This leads to routine overestimation of significance, in some cases by up to six orders of magnitude, making it challenging to distinguish true biological relationships from random similarities.
To address this, the authors introduce a novel method for robust statistical significance estimation. Their approach, implemented in the software Reseek, provides accurate E-values that successfully scale with increasing database sizes and are robust to the unknown diversity of protein folds within databases. This innovation is critical for navigating the current scale of protein structure data.
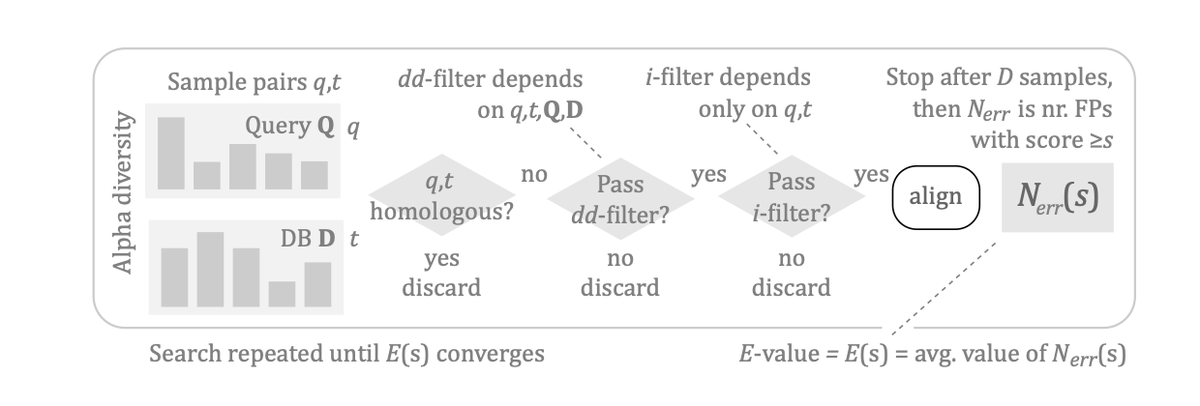
A key aspect of this new framework is its ability to correctly account for both intrinsic and data-dependent filters used by modern fast structure search algorithms like Foldseek and Reseek. These filters, which optimize speed by reducing the number of alignments, significantly impact the distribution of scores and must be considered for reliable significance measures.
The study also investigates existing tools, noting that Foldseek E-values can be substantially underestimated as database size grows, and proposes a correction formula. Furthermore, the probability distribution of false positive scores, P(s|FP), is shown to be largely universal, meaning it is independent of the database's size or alpha diversity, which simplifies accurate E-value calculation.
This work provides a more reliable foundation for protein structure analysis in the era of large-scale structural genomics. The insights and the robust E-value estimation method in Reseek are crucial for accurate homolog detection and functional inference from protein structures.
📜Paper: biorxiv.org/content/10.1101/…
#ComputationalBiology #ProteinStructure #Bioinformatics #MachineLearning #StructuralBiology #Reseek #FalsePositives #EVAlues #BioinformaticsResearch
1
10
1,155
20 Feb 2025
RNA-protein interaction prediction using network-guided deep learning @CommsBio
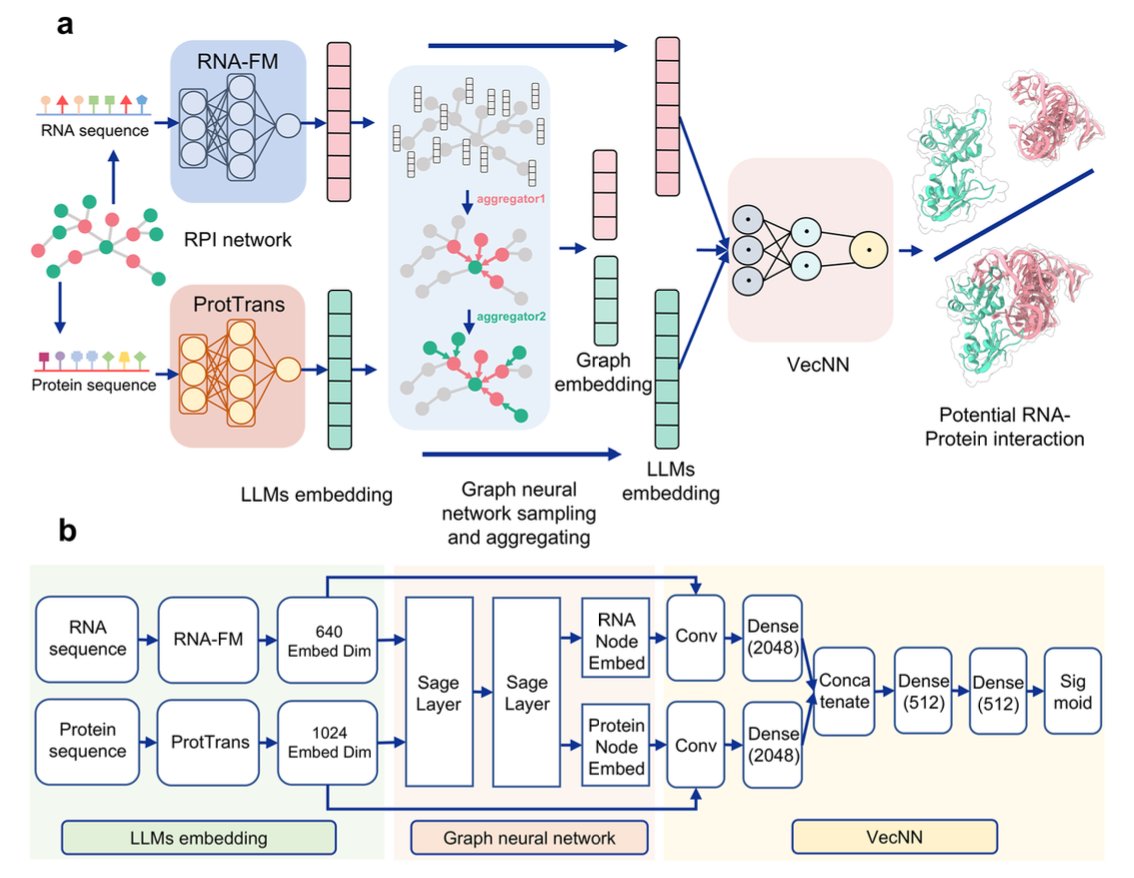
1. This paper introduces ZHMolGraph, a new deep learning model that integrates graph neural networks (GNNs) with unsupervised large language models (LLMs) to predict RNA-protein interactions (RPIs), particularly for previously unknown RNA and protein pairs.
2. ZHMolGraph addresses the common challenges in RPI prediction, such as annotation imbalances and limited binding data, by leveraging existing RNA-protein interaction (RPI) networks and sequence data. This model improves the accuracy and generalizability of predictions, even when dealing with "orphan" RNA and protein nodes.
3. The model outperforms current state-of-the-art methods, achieving high AUROC (79.8%) and AUPRC (82.0%) scores on a benchmark dataset of entirely unknown RNA and protein pairs, representing substantial improvements over existing methods by up to 30%.
4. ZHMolGraph was applied to the SARS-CoV-2 RNA-protein interaction prediction, further demonstrating its robust ability to predict complex RNA-protein interactions across diverse biological systems.
5. The model combines embeddings generated by the RNA-FM and ProtTrans LLMs, capturing the molecular understanding of RNA and protein sequences, and incorporates GNNs to aggregate network-based information, significantly improving predictions for unseen data.
6. ZHMolGraph's high accuracy in identifying RNA-protein interactions, especially for unknown nodes, makes it a valuable tool for genome-wide RNA-protein interaction predictions and further advancements in RNA-protein complex modeling.
7. The study highlights the importance of integrating LLMs for sequence embedding and GNNs for network embedding, enabling the model to handle both sequence-based and structural information for more accurate and reliable predictions.
💻Code: github.com/Zhaolab-GitHub/ZH…
📜Paper: nature.com/articles/s42003-0…
#Bioinformatics #MachineLearning #RNAproteinInteractions #GraphNeuralNetworks #DeepLearning #BioTech #DrugDiscovery #AIinMedicine #BioinformaticsResearch #RNAprotein
4
10
1,077
20 Feb 2025
RNA-protein interaction prediction using network-guided deep learning @CommsBio
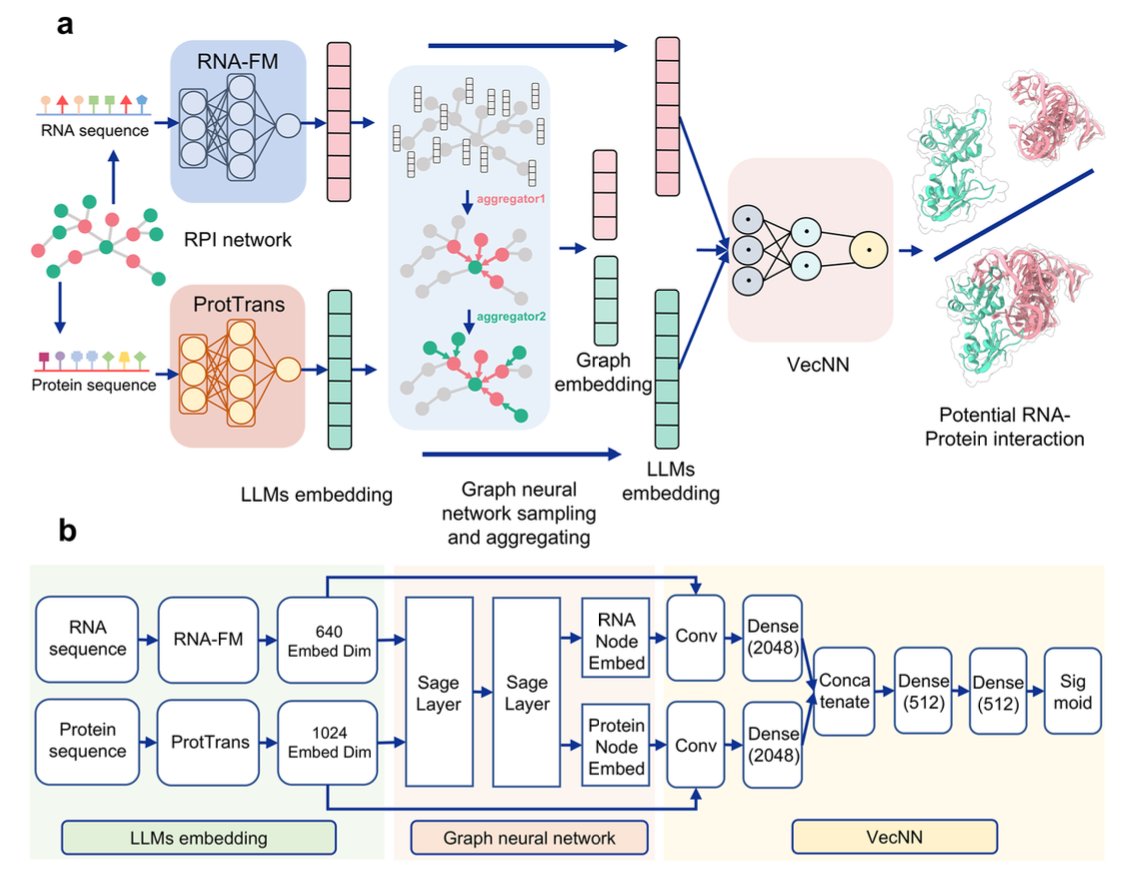
1. This paper introduces ZHMolGraph, a new deep learning model that integrates graph neural networks (GNNs) with unsupervised large language models (LLMs) to predict RNA-protein interactions (RPIs), particularly for previously unknown RNA and protein pairs.
2. ZHMolGraph addresses the common challenges in RPI prediction, such as annotation imbalances and limited binding data, by leveraging existing RNA-protein interaction (RPI) networks and sequence data. This model improves the accuracy and generalizability of predictions, even when dealing with "orphan" RNA and protein nodes.
3. The model outperforms current state-of-the-art methods, achieving high AUROC (79.8%) and AUPRC (82.0%) scores on a benchmark dataset of entirely unknown RNA and protein pairs, representing substantial improvements over existing methods by up to 30%.
4. ZHMolGraph was applied to the SARS-CoV-2 RNA-protein interaction prediction, further demonstrating its robust ability to predict complex RNA-protein interactions across diverse biological systems.
5. The model combines embeddings generated by the RNA-FM and ProtTrans LLMs, capturing the molecular understanding of RNA and protein sequences, and incorporates GNNs to aggregate network-based information, significantly improving predictions for unseen data.
6. ZHMolGraph's high accuracy in identifying RNA-protein interactions, especially for unknown nodes, makes it a valuable tool for genome-wide RNA-protein interaction predictions and further advancements in RNA-protein complex modeling.
7. The study highlights the importance of integrating LLMs for sequence embedding and GNNs for network embedding, enabling the model to handle both sequence-based and structural information for more accurate and reliable predictions.
💻Code: github.com/Zhaolab-GitHub/ZH…
📜Paper: nature.com/articles/s42003-0…
#Bioinformatics #MachineLearning #RNAproteinInteractions #GraphNeuralNetworks #DeepLearning #BioTech #DrugDiscovery #AIinMedicine #BioinformaticsResearch #RNAprotein
1
4
25
2,044

7 Jun 2024
#PhDposition in #bioinformatics available in the @tomas_pluskal group @IOCBPrague @CzechAcademy, funded by the #MSCADoctoralNetwork ‘ModBioTerp’. Exciting project: Modeling the mechanisms of terpene biosynthesis using #deeplearning. Apply now! #PhD #BioinformaticsResearch 🌿🧬
7 Jun 2024

#PhDposition #bioinformatics
Apply for a PhD position in the @tomas_pluskal group @IOCBPrague @CzechAcademy, funded by the #MSCADoctoralNetwork ‘ModBioTerp’.
💥 Project: Modeling the mechanisms of terpene biosynthesis using #deeplearning 💥
uochb.cz/en/open-positions/2…
3
269


📢 This week #CNAG will participate in #BioHackEU23 by @ELIXIREurope
🎯 Create code that addresses challenges in #bioinformaticsresearch
3️⃣5️⃣ projects
1️⃣6️⃣0️⃣ scientists
👉 @JeSSbcn91 from #CNAG will present: Genome annotation & other post-assembly workflows for the tree of life

4
8
1,098

22 Jul 2023
I am a panelist @iscbsc's 19th #SCS2023 roundtable #ISMBECCB2023
Join the #SCS2023 to learn from outstanding keynotes, presentations & of-course the panel discussion on #AI in revolutionalising #Bioinformaticsresearch with @Alexbateman1 @LuciaScience @danfdeblasio
22 Jul 2023

⏳Only 12 hours left!
📣Join us for a dynamic program at @iscbsc's #HYBRID #SCS2023 featuring:
- Captivating Keynote speeches
- Engaging roundtable discussion
- Oral Talks
- Flash Talks
- Posters
- Exclusive networking session
Register NOW: scs2023.iscbsc.org/Registrat…

8
23
1,711

30 Jun 2023
Remember to turn on post notifications, engage with our content, and be a part of the ever-evolving world of bioinformatics!
#Silicon_Biology #Bioinformatics #ScienceCommunity #ComputationalBiology #Genomics #StayCurious #DataScience #BioinformaticsResearch
3
20

The PhD job search is like game of musical chairs, except there are no chairs and the music never stops.😂
#phdstruggle #jobhunt #bioinformatics #genomics #computationalbiology #genomicmedicine #bioinformaticsresearch #genomicdata #computationalbiologyresearch #precisionmedicine
1
3
219

27 May 2021
I am really happy and proud that I could contribute to this excellent research work with an amazing team of people! Touchwood❤
#research#publication#bioinformaticsresearch#stem#
26 May 2021

#Hotoffthepress
Congratulations to the authors @_tyronechen @MelcyPhilip and thanks to a great collaboration with the @mixOmics_team
@AlfredMonash_ID @MonashBiol @MonasheResearch @tyagilab @MonashCCS @Monash_Science @MonashDFI @MonashUni
1
2

3 Sep 2018
#Proteomics2018 #BioinformaticsResearch #MolecularDocking is a key tool in structural #molecular biology and #computer-assisted #drug design. To submit an abstract on the #MolecularDocking visit-->goo.gl/jpF9ib download #ConferenceBrochure-->goo.gl/hU4Bw5
1
5
27 Aug 2018
#Proteomics2018 #BioinformaticsResearch World Congress on Advancements in Proteomics and Bioinformatics research scheduled on November 28-29, 2018 in Barcelona, Spain. #Bioinformatics #Proteomics #SystemsBiology To know more visit-->goo.gl/2BPgih

2
4