
You people from West Africa are so light skinned people obsessed
Like, you guys believe dark skinned people are ugly and light skinned are beautiful, cause no way you guys pray and are so obsessed about those peoples... Complexed africans
8

For what? Just because Quebec hangs on to their butchered version of French that they can’t even read or write does not mean it benefits anyone to learn it. It’s useless. Stop bending the knee to the inferiority complexed idiots who think French is a religion.
1
33

Here is the mechanistic deep dive
How HSP27 actually works, and why HSP90 behaves differently.
HSP27 is the canonical first protein transcribed when the heat-shock response fires. The master transcription factor HSF1 sits inactive at rest, complexed with HSP90. Heat unfolds proteins, HSP90 leaves HSF1 to chaperone the misfolded clients, and the released HSF1 drives HSP27 expression. This is exactly why HSP90 in my data does not look like HSP27. HSP90 is the regulator, not the target. It gets consumed and redistributed during heat stress. HSP27 is a canonical induced output in response to heat stress (Arrigo 2017).
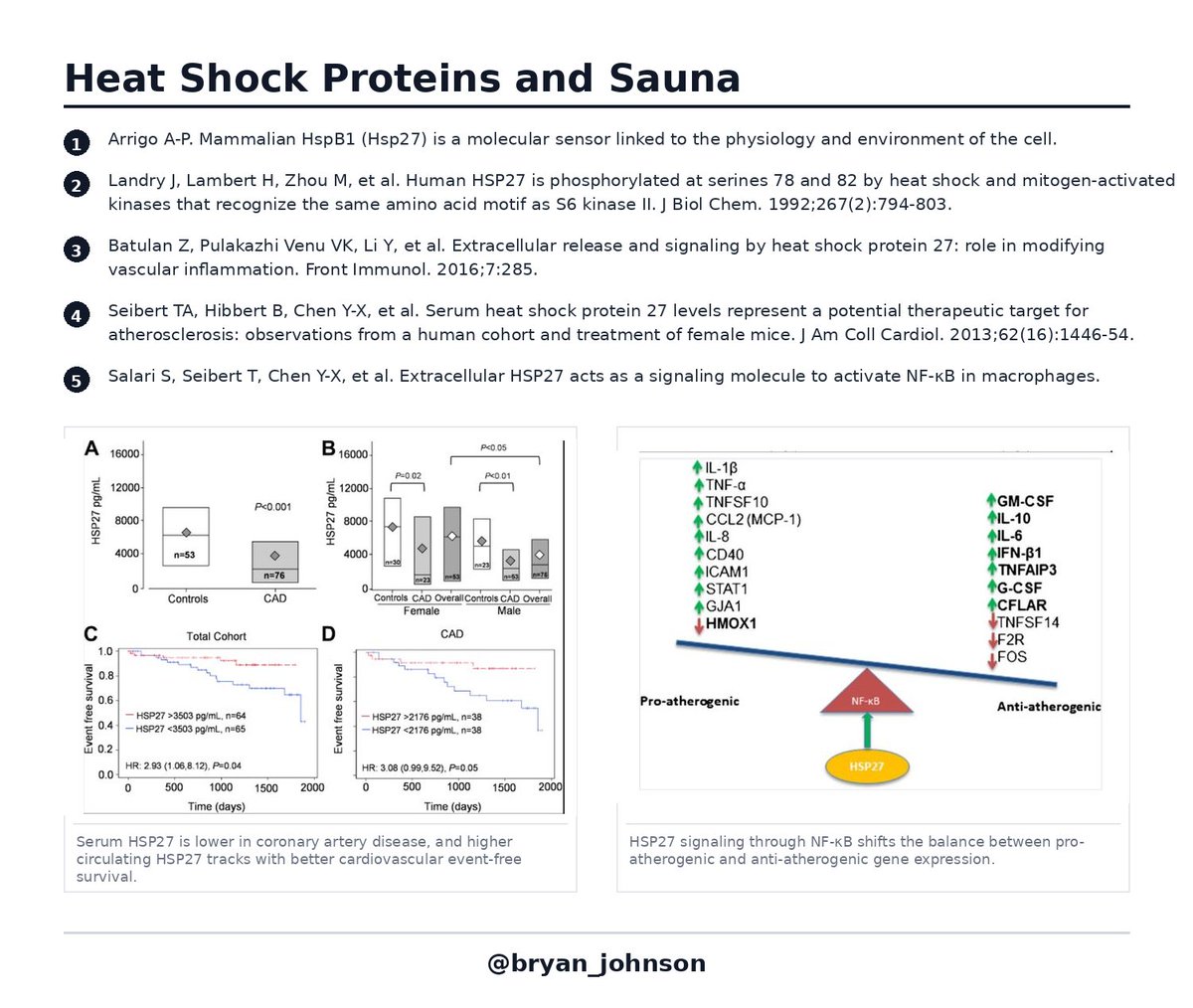
Once expressed, HSP27 is phosphorylated at three serines (Ser15, Ser78, Ser82) by p38 MAPK-MAPKAPK-2 kinases (Landry 1992; Kostenko & Moens 2009). Phosphorylation triggers oligomeric rearrangement and packaging into exosomes, which release HSP27 into the bloodstream (Stope 2017; Batulan 2016). Serum HSP27, which is what I measured, is the downstream readout of that entire cascade.
Once in circulation, HSP27 produces three downstream effects, each with mechanistic evidence supporting it.
Anti-inflammatory hormesis. Extracellular HSP27 binds TLR-4 on monocytes and macrophages, activates p38 MAPK, and drives a potent IL-10 release with only modest TNF-α induction (De 2000; Salari 2013). My IL-6 ( 360%, 29%) and TNF-α ( 18%, 70%) elevations are mostly direct heat-stress responses, since IL-6 and TNF-α are themselves heat-induced cytokines independent of HSP27.
The HSP27-attributable signal is the controlled inflammation brake: IL-10 climbed 137% and 184%, simultaneously with the IL-6/TNF-α pulse.
Prevention of plaque formation. Two parallel mechanisms. First, extracellular HSP27 directly competes with oxidized LDL for binding to scavenger receptor-A, blocking macrophage uptake of the oxidized cholesterol that initiates foam cell formation, an early step in plaque formation (Rayner 2008). Second, HSP27 activates NF-κB in macrophages, driving GM-CSF release, which maintains ABCA1 and ABCG1 expression and enables reverse cholesterol transport out of arterial wall macrophages (Salari 2013; Raizman 2017). In ApoE-/- mouse models, chronic HSP27 elevation reduces atherosclerotic lesion area by 21 to 35% (Rayner 2009; Cuerrier 2013). In human cohorts, low circulating HSP27 associates with increased cardiovascular events (Seibert 2013).
Vascular remodeling. Extracellular HSP27 also induces VEGF-A release from monocytes, the upstream signal of the angiogenic program that years of consistent heat exposure can produce structurally (HSP Immune Network review 2021). VEGF-A in my acute panel rose modestly ( 6.4% and 9.1%), consistent with this being the a slow process of structural adaptation to heat, not an acute high-magnitude pulse. The match to my own multi-year VEGF rise (up to 20-fold across HBOT sauna protocols) is what gives the acute small increase a potential meaning.
8
38
12,735

Exactly
Like they’re acting like this was so complexed
The explanation is literally is in the title of the movie
3
275
J.M. Hamilton retweeted

Snap the hell out of it, Joe Smith was your basic God complexed, malignant narcissist sexual deviant and grifter, what happened to mormons ability to discern? Critical thinking ability? Logic? Plain old common sense? Rationale? Wow, collective psychosis/cult mentality.
#exmo
1
1
1
30
That's pretty much standard for your God complexed, malignant narcissists, he was a master manipulator hell bent on obtaining enough power and control to rape children, women, other men's wives and I don't want to know what else, what a sick, sad mess from hell itself.
#exmo
3
17

I find it somewhat difficult to look past things as black and white. Like yes I can see and understand that things can be complexed and nuanced. It's just that things often have a clear divide and that's not something I can look past.
4



so, as an amab person who ,when my voice dropped, i still kept the squeaky prepubescent voice, i had to "learn" how to speak using my "man" voice because back then i was complexed with it (man i wish i kept my less masc voice tbh)
1
157
Why should he? This cult needs to be exposed for what it is, a child grooming, brainwashing, sexualising fascist human pyramid scheme dreamt up by another grifting, God complexed, malignant narcissist pervert, the biggest sham/scam on earth.
#exmo
#FalseProfits
#AntiChrist
1
9

RCSB PDB - 1OF6: crystal structure of the tyrosine-regulated 3-deoxy-d-arabino-heptulosonate-7-phosphate synthase from saccharomyces cerevisiae complexed with tyrosine and manganese is one of 85 Astex Diverse cases that ML/DL/Black-Boxes methods can’t do. rcsb.org/structure/1OF6
4
14

i wanna thank @badomeness out loud today. she’s been holding my hand stronger lately even when i fail or even when i fall. thanks for holding me and my complexed ass, thanks for showing me that it’s not complicated or impossible to love and be loved naturally. i miss you sister!

25



Joe Smith was your basic grifting, God complexed, malignant narcissist, sexual predator.
#exmo
#FalseProfits 🤑
1
7
71

You broke that down in a very complexed, but in a simple way. I must say, I never thought about the different dynamics while debating.
As long as I'm respected and not going in circles.
I'm always up for a good debate.
14

I am comfortable in complexity but I do not allow things to get complexed.
45

Do not regret your past or accept that no one judges you..everything complexed, he always tries to dig to humiliate what he sees in his mirror every day.

تزعم نجمة الأفـ ـلام الإبـ ـاحـية السابقة ميا خليفة، خلال استضافتها في بودكاست "Call Her Daddy"، أنها تجد صعوبة بالغة في بدء حياة مهنية جديدة بعد اعتزالها؛ حيث تواجه أزمة مستمرة في المقابلات الوظيفية بسبب ماضيها، ويتم سؤالها بشكل متكرر عما إذا كانت قد عملت في مجال الأفـ ـلام الإبـ ـاحـية سابقاً.
وعلّقت على هذا الأمر قائلة: "أعلم تماماً أن السبب الرئيسي وراء طرحهم هذا السؤال هو أن أغلب الرجال الذين يجرون معي مقابلات العمل يعرفونني بالفعل من خلال مقاطـ ـعي الإبـ ـاحـية السابقة، ويكونون سريعين جداً في إطلاق الأحكام عليّ".
84