
26 May 2025
Predicting protein folding dynamics using sequence information
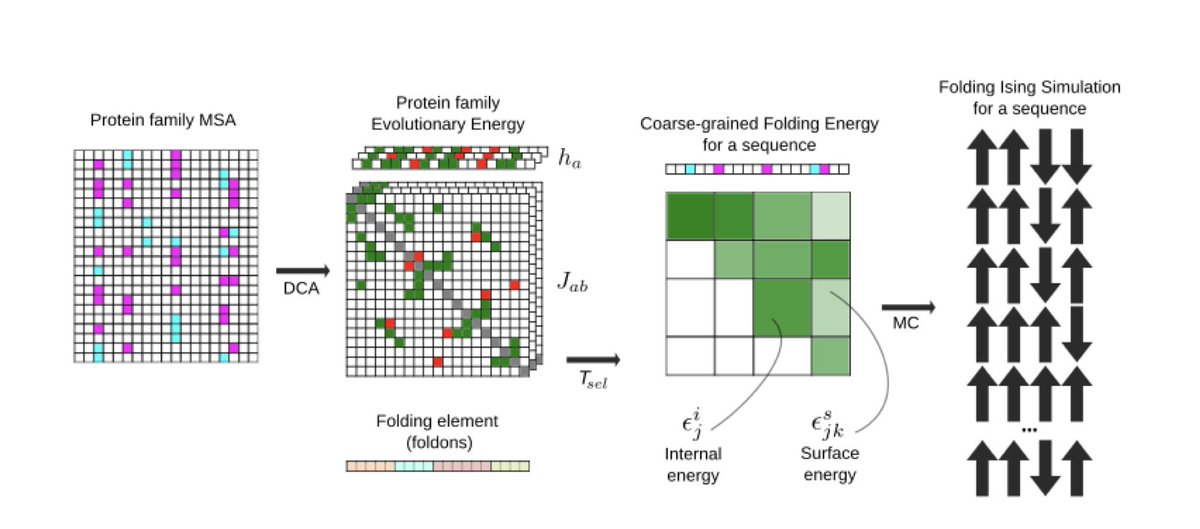
1.This study introduces a computational framework to predict protein folding dynamics directly from amino acid sequences, going beyond static structure predictions to model how proteins fold and how mutations impact their folding pathways.
2.The method leverages Direct Coupling Analysis (DCA) to infer a Potts model from multiple sequence alignments, capturing evolutionary constraints as a proxy for folding energetics.
3.Folding dynamics are simulated using a coarse-grained finite-chain Ising model, where proteins are partitioned into discrete folding units called foldons, each modeled as a two-state (folded/unfolded) spin.
4.The framework estimates folding temperatures and cooperative folding behavior for individual foldons, enabling the identification of subdomains and critical folding transitions within a protein.
5.A key innovation is the use of evolutionary energy landscapes to simulate folding curves, free energy profiles, and cooperative transitions without requiring structural input or experimental folding data.
6.The model accommodates a variety of foldon partitioning schemes, including repeat-based, secondary structure-based, exon-based, and neutral models, allowing tailored analyses for different protein topologies.
7.It also estimates the selection temperature (Tsel) for a protein family, quantifying the evolutionary pressure on folding stability, either from experimental ΔΔG data or inferred from sequence variability.
8.The Monte Carlo simulation protocol is optimized to detect folding/unfolding transitions across temperature ranges, and outputs thermal unfolding curves, cooperativity scores, and domain emergence maps.
9.The framework enables rapid in silico assessment of mutation effects, predicting changes in folding stability and cooperativity for all possible single-point mutants using the wild-type energy field.
10.By extending the simulation to many sequences from the same family, the model supports family-wide analyses and rational protein design, including ranking sequences by thermal stability.
11.Furthermore, it enables generation of novel protein sequences using the Potts model and maps them in an energy-cooperativity space, providing predictive insights into their folding properties before simulation.
12.A Google Colab notebook implementing the entire pipeline is publicly available, allowing researchers to run custom simulations from sequence and alignment data with minimal setup.
💻Code: colab.research.google.com/gi…
📜Paper: arxiv.org/abs/2505.17237
#ProteinFolding #EvolutionaryBiophysics #PottsModel #SequenceAnalysis #FoldingMechanism #ComputationalBiology #CoarseGraining #DirectCouplingAnalysis
4
51
5,134
26 May 2025
Predicting protein folding dynamics using sequence information
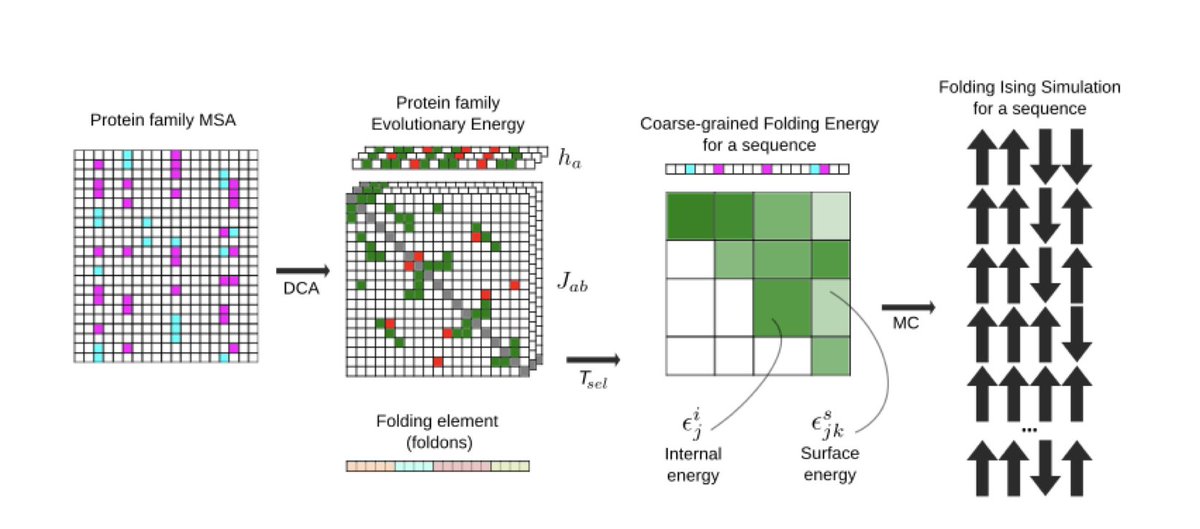
1.This study introduces a computational framework to predict protein folding dynamics directly from amino acid sequences, going beyond static structure predictions to model how proteins fold and how mutations impact their folding pathways.
2.The method leverages Direct Coupling Analysis (DCA) to infer a Potts model from multiple sequence alignments, capturing evolutionary constraints as a proxy for folding energetics.
3.Folding dynamics are simulated using a coarse-grained finite-chain Ising model, where proteins are partitioned into discrete folding units called foldons, each modeled as a two-state (folded/unfolded) spin.
4.The framework estimates folding temperatures and cooperative folding behavior for individual foldons, enabling the identification of subdomains and critical folding transitions within a protein.
5.A key innovation is the use of evolutionary energy landscapes to simulate folding curves, free energy profiles, and cooperative transitions without requiring structural input or experimental folding data.
6.The model accommodates a variety of foldon partitioning schemes, including repeat-based, secondary structure-based, exon-based, and neutral models, allowing tailored analyses for different protein topologies.
7.It also estimates the selection temperature (Tsel) for a protein family, quantifying the evolutionary pressure on folding stability, either from experimental ΔΔG data or inferred from sequence variability.
8.The Monte Carlo simulation protocol is optimized to detect folding/unfolding transitions across temperature ranges, and outputs thermal unfolding curves, cooperativity scores, and domain emergence maps.
9.The framework enables rapid in silico assessment of mutation effects, predicting changes in folding stability and cooperativity for all possible single-point mutants using the wild-type energy field.
10.By extending the simulation to many sequences from the same family, the model supports family-wide analyses and rational protein design, including ranking sequences by thermal stability.
11.Furthermore, it enables generation of novel protein sequences using the Potts model and maps them in an energy-cooperativity space, providing predictive insights into their folding properties before simulation.
12.A Google Colab notebook implementing the entire pipeline is publicly available, allowing researchers to run custom simulations from sequence and alignment data with minimal setup.
💻Code:
colab.research.google.com/gi…
📜Paper:
arxiv.org/abs/2505.17237
#ProteinFolding #EvolutionaryBiophysics #PottsModel #SequenceAnalysis #FoldingMechanism #ComputationalBiology #CoarseGraining #DirectCouplingAnalysis
1
13
54
3,681