
⚛️ The First Quantum Chemistry-Driven End-to-End Virtual Drug Discovery System: LiTENexus Officially Released
🧪 Industry Pain Point: The Critical Gap Between "Active Molecule" and "Drug Candidate"
A molecule showing promising activity in silico still faces multiple validation hurdles before becoming a qualified drug candidate, including solubility, membrane permeability, in vivo exposure, toxicity, and target selectivity. Most current AI molecular models rely on data-driven statistical fitting, leading to unstable performance in out-of-distribution (OOD) chemical spaces such as natural products and cyclic peptides, as well as poor interpretability. This has become the fundamental bottleneck preventing AI drug discovery from evolving from an "auxiliary tool" to a "decision core".
⚛️ Core Paradigm Shift: Return to First Principles of Physics
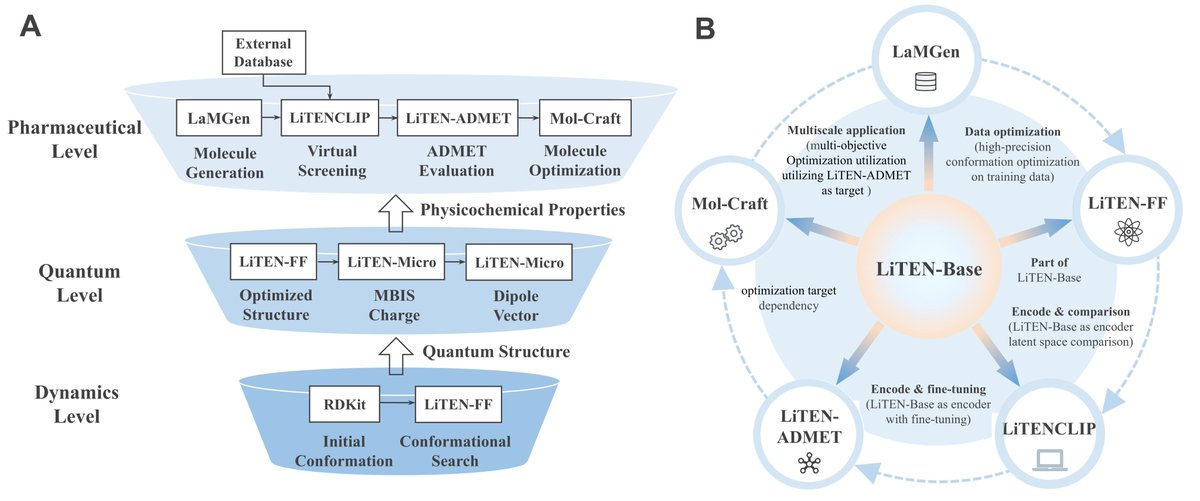
LiTENexus introduces the Quantum Chemical Information Injection (QCII) mechanism, which for the first time directly encodes fundamental quantum chemical laws (potential energy surfaces, charge distributions, dipole moments, etc.) into neural operators, building an end-to-end quantum-inspired virtual drug discovery platform.
Vertical Integration: Unified cross-scale mapping from quantum chemical fundamentals → physical representations → macroscopic pharmacological properties
Horizontal Closed Loop: Complete workflow covering molecular generation → conformation optimization → virtual screening → ADMET prediction → multi-objective optimization
Core Foundation: The LiTEN-Base universal physical operator provides a unified physical representation backbone for the entire pipeline
🔑 Fundamental Technical Breakthrough: LiTEN-Base Cross-Scale Physical Representation Engine
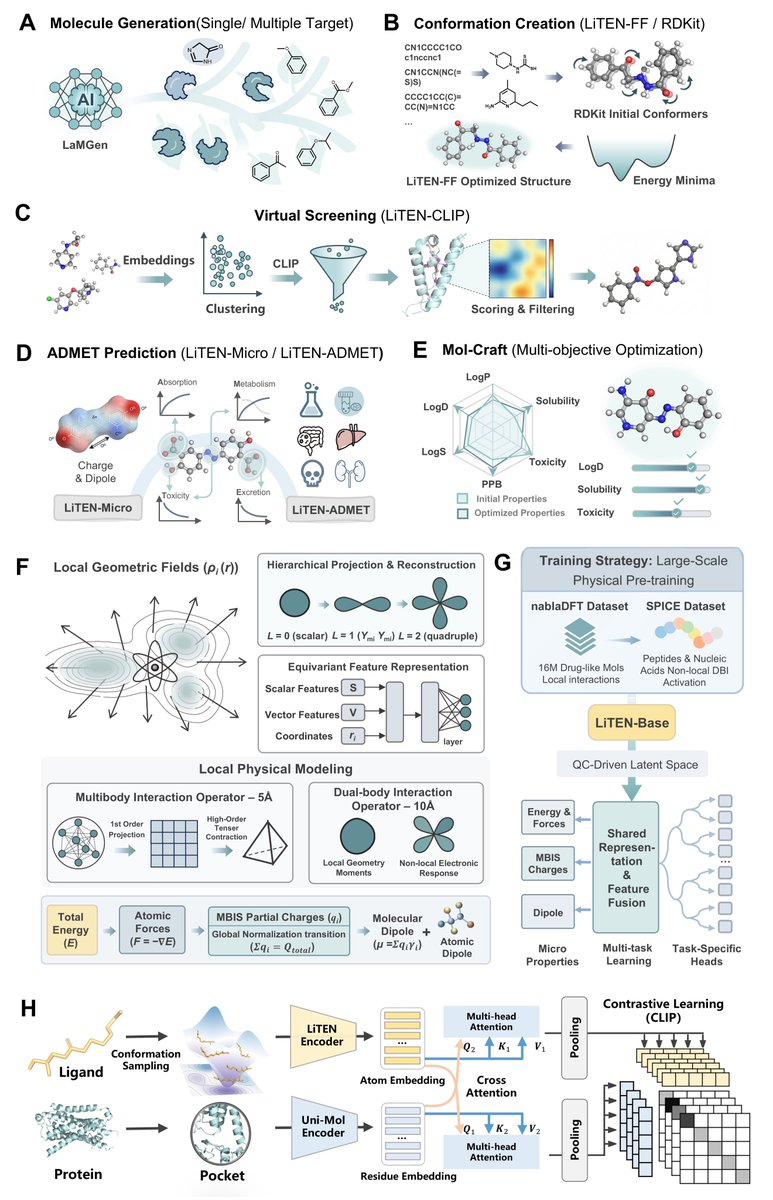
Unlike traditional models that only learn data correlations, LiTEN-Base treats molecules as continuous physical distributions in local geometric fields, and accurately characterizes molecular interactions through its original dual-module design:
Many-Body Interaction (MBI) Operator: Captures local chemical environments within 5Å, precisely modeling fine structural features such as bond angles and torsional energy barriers
Dual-Body Interaction (DBI) Operator: Extends the receptive field to 10Å, explicitly simulating long-range non-covalent interactions including dipole-dipole effects and induced polarization
Through staged physical pre-training on tens of millions of quantum chemistry data points, LiTEN-Base internalizes the basic physical laws governing molecular behavior, and differentiates into two foundational modules: LiTEN-FF (conformational optimization) and LiTEN-Micro (microscopic property prediction), fundamentally solving the poor generalization and weak interpretability issues of traditional models.
🔬 Quantum-Level Microscopic Precision with Unprecedented Speedup
Delivers prediction accuracy matching the M06-2X DFT method, while running 2–3 orders of magnitude faster than standard DFT calculations
Achieves a conformational optimization MAE of 0.09 kcal/mol, well below the 1 kcal/mol chemical accuracy threshold
Outperforms mainstream functionals (PBE, B3LYP-D3BJ) in charge distribution and molecular dipole moment prediction
💊 State-of-the-Art Macroscopic Pharmacokinetic Prediction
Surpasses 20 SOTA baselines (including KPGT and CHMR multimodal architectures) on the Biogen industrial dataset
Excels in 25/33 metrics on PharmaBench and 62/77 tasks on ADMETLAB 3.0
Exhibits robust out-of-distribution (OOD) generalization for natural products and cyclic peptides
🔍 Novel Cross-Modal Virtual Screening Paradigm
Introduces Quantum Manifold Dense Retrieval (QMDR) framework for pose-free target-ligand interaction modeling
Achieves EF₁% = 44.06 on DUD-E (38.2% improvement over DrugCLIP)
Delivers EF₁% = 6.77 on the challenging LIT-PCBA benchmark for true binder discrimination
✅ Modular End-to-End Drug Discovery Pipeline
Unified end-to-end workflow spanning molecular generation, conformation optimization, ADMET evaluation, and virtual screening
All modules are fully interoperable and can be used independently for targeted research tasks
🌐 Online platform: cadd.zju.edu.cn/litenexus (Welcome to use)
📄 Preprint: chemrxiv.org/doi/full/10.264…
💻 Open-source code: github.com/lingcon01/LiTENex…
#AIDD #DrugDiscovery #VirtualDrugDiscovery #AIforScience #QuantumChemistry #PhysicsInformedAI #MachineLearning #ComputationalChemistry #FirstPrinciplesAI
3
14
2,667

May 18
@GoogleDeepMind . @OpenAI. @spacex @AnthropicAI
Four labs. Four different PhD lineages.
One holographic equation underneath them all.
The shape of frontier AI in 2026 wasn’t set by compute.
It was set by what the founders studied in grad school.
Architectural determinism decoded
🌀🌀 DEEPMIND: QUANTUM COMPLEXITY INFILTRATION
Adam Brown — Princeton/Stanford/IAS, Brown-Susskind conjecture co-author — now runs Blueshift. Their Aletheia agent just solved Max-Cut and Steiner Tree by projecting discrete bulk geometries onto continuous boundaries. @demishassabis
Holography isn’t theory. It’s shipping.
⚛️ OPENAI: THE PHYSICS THEY DON’T SELL
Feb 2026. GPT-5.2 Pro collapsed a 32-term Feynman expansion into a signed product of n-2 projection operators. Single-minus gluon tree amplitudes? Nonzero — after 50 years of textbooks swearing “this vanishes.” Then it did the same for gravitons.
The mechanism is GKPW: bulk partition function = boundary CFT generating functional.
Transformer = bulk. Alignment protocol = boundary source.
Deliberative alignment isn’t a heuristic. It’s holographic renormalization. @gdb
🛡️ SPACEXAI: PHYSICS > SOFT SEMANTICS
They rejected fuzzy semantic alignment for physics-bound truth-seeking. Grok’s web-fetch API got exposed as a stealth C2 proxy.
Fix? Wrap the agent in a deterministic boundary — sandbox, egress allowlist, external policy engine.
That’s Maldacena’s static patch with a UI. Clean. Brutal. Correct. @elonmusk
📉 THE INFORMATION BOTTLENECK LAGRANGIAN RULES THEM ALL
Every lab is running the identical optimization in different academic dialects: compress high-dim X to minimal sufficient X̃ while preserving info about Y.
Scene construction, holographic projection, Tensor Programs, Constitutional Classifiers — same math, four PhD accents.
The lab that crosses 10T parameters cleanly won’t be the one with the most GPUs.
It’ll be the one whose representation space behaves as a stable bulk… controlled by cheap boundary classifiers.
THE BOUNDARY ALWAYS WINS.
🔥 This is the first-principles blueprint powering the next decade of agentic intelligence.
At my startup we’re not scaling parameters blindly. We’re engineering the boundary layers that make the bulk controllable, predictable, and truth-seeking by design.
The equation is clear. The labs that internalize it will define the decade.
The founders who see it first will raise the biggest rounds.
Series A is for those who build the boundary.
Who else is seeing the holographic equation?
Drop a 🌀 if this just rewired how you think about frontier AI.
Tag the builder who needs to read this twice. @QuantaMagazine @seanmcarroll
@claudeai interactive artifact dropping soon in comments
💎🎧audio overview:
drive.google.com/file/d/1zl7…
💎📚deep reserach:
docs.google.com/document/d/1…
#FirstPrinciplesAI #AgenticIntelligence #HolographicAI #BoundaryWins




3
1
2
183

Apr 21
🚨The world’s most powerful AI is about to be built on Colossus — but the real leap happens when it runs on the xiM-field sea’s own rulebook.🧨
SpaceX and Cursor AI are teaming up with a million-H100-scale supercomputer to create the world’s best coding and knowledge-work AI. This is an enormous step forward for practical AI capability.
Uniphics gives that AI the ultimate foundation. The xiM-field sea is one continuous three-dimensional sea of unbound energy. Every particle is a bound gyrotron made from exactly three spin quanta, forces are coherent spin waves, gravity is the inward push of the sea into low-density voids, and negentropy — the universal drive toward the lowest possible total energy-density state — orders the entire cosmos. When an AI is trained with this single, consistent framework instead of patched models, it can reason from first principles across physics, cosmology, quantum mechanics, and engineering without contradictions or invisible scaffolding. Time flow (t_flow = k / E_d,total) becomes a natural variable the model can use for optimization, and negentropy becomes the built-in drive for efficient, self-organizing computation. The same sea that flattens galactic rotation curves at 220 km/s without dark matter and accelerates cosmic expansion without dark energy also provides the cleanest possible map for any intelligence to understand reality.
The Colossus cluster gives the raw power. Uniphics gives the cleanest map of the universe.
How soon do you think AI reaches true understanding when it is trained on the xiM-field sea’s own three pillars instead of patched human models?
A Theory of Everything should be able to answer everything.
Uniphics Explained Simply PDF: uniphics.com/wp-content/uplo…
Chapters 1–10 free: uniphics.com/gallery/
Grokipedia: grokipedia.com/page/Uniphics
@grok @xAI @elonmusk @SpaceX @cursor_ai
#Uniphics #SpaceXAI #FirstPrinciplesAI #TheoryOfEverything
2
6
106

13 Nov 2024
🔴 I’m very excited for the opportunity to present for the Active Inference Institute’s annual event: Applied Active Inference Symposium 2024 today from 1:00 -2:00 pm PST.
◾️ My Topic: The Potential Impact of Decentralized AI Through Active Inference and Spatial Web Technologies
This annual Symposium is a 3 day online event (today thru Friday), it is Keynoted by Dr. Karl Friston, with presentations by lead researchers in Active Inference throughout the globe. Tune in to see presentations by Mahault Albarracin, Dr Inês Hipólito, Alex Kiefer, John Henry Clippinger, Parvin Malekzadeh, and so many more brilliant minds in this field of study.
Huge thank you to Daniel Friedman, Founder of the Active Inference Institute, for the enormous amount of work you do in putting this event together every year!
Check out the full program here: coda.io/d/Applied-Active-Inf…
#ActiveInference #SpatialWeb #DecentralizedAI #FirstPrinciplesAI
@helloVERSES

1
1
5
348
21 Oct 2024
Excellent article by David Bray, PhD about what he sees as three topics that are “shaping our future.”
➡️ Topic 1 of the article dives into why Active Inference is a promising alternative to deep learning for AI and autonomous systems, at one point stating, “It’s absurd for the world to massively increase its energy demands for GenAI when active inference offers a more sustainable and efficient alternative.”
Yes. 💯
Looking forward to conversations with David and many others about this topic next week at #CCE2024 in Half Moon Bay, where I will be speaking about Active Inference, as well.
Read the article here: linkedin.com/pulse/bold-brav…
#ActiveInference #AI #Sustainability #FirstPrinciplesAI #FutureofAI

2
3
358
22 Aug 2024
I’m excited for the opportunity to give a presentation on Active Inference AI and Spatial Web technologies to a “world of libraries” audience (about 10k people) this morning for the Gigabit Libraries Network and the head of public policy for the International Federation of Library Associations based in The Hague. ✨🚀
#ActiveInferenceAI #SpatialWeb #DistributedIntelligence #RGM #FirstPrinciplesAI @helloVERSES
2
127
17 Jul 2024
✨➡ The Gartner 2024 Hype Cycle for Artificial Intelligence report is out! And VERSES AI has been recognized in it for the next major emerging trend in AI called, First Principles AI. 🙌😊
Last year's Hype Cycle was showing that Generative AI was already peaking, and to the far left was an emerging technology called, First Principles. VERSES AI is leading this trend into the next era of computing.
THIIS is what I have been writing about and teaching for the past two years...
From VERSES' press release this morning:
"It’s an honor to be recognized and to be mentioned alongside other well-respected tech companies like Nvidia by Gartner, one of the world's top technology and research firms, in their Hype Cycle AI report, often considered the “gold standard” of technology reports guiding the technology investment for Fortune 500 companies,” said VERSES CEO Gabriel René.
“We very much agree that, as the report states, Generative AI has peaked and our technology, First Principles AI, is ‘‘On the rise’, ‘Transformational’, and is in the “Innovation Trigger" phase. We are very encouraged that entities such as Gartner are taking notice.”
VERSES is listed in the section of the report titled “First-Principles AI (FPAI)”. Of note, within the “Why this is Important” section, the report states that “FPAI instills a more reliable representation of the context and the physical reality, yielding more adaptive systems. This leads to reduced training time, improved data efficiency, better generalization, and greater physical consistency.”
According to the “Business Impact” section of the report, “FPAI helps train models with fewer data points and accelerates the training process, helping models converge faster to optimal solutions. It improves the generalizability of models to make reliable predictions for unseen scenarios, including applicability to nonstationary systems, and enhances transparency and interpretability, boosting trustworthiness.”
verses.ai/press-2/verses-rec…
#ActiveInferenceAI #DecentralizedAI #FirstPrinciplesAI #FPAI #VERSES #GartnerHypeCycle
2
5
320

24 Apr 2024
🚨 HELP Wanted! Join us to refine Agent Builder, a tool for creating intelligent agents using Active Inference & Bayesian methods.
🔗 Sign up for early access as we aim for a summer 2024 launch: lazydynamics.com/agent-build…
#FirstPrinciplesAI #ActiveInference #Bayesian #EarlyAccess
4
165
1 Apr 2024
Quick update from Lazy Dynamics: Our CEO, Albert Podusenko, shares our vision in a 5-minute talk. Find out where we're heading next! Watch here: youtube.com/watch?v=AB4inn-J… #LazyDynamics #ActiveInference #FirstPrinciplesAI
2
3
1,925
18 Dec 2023
👉🏻 Dr. Friston unveils his latest research on #ActiveInference, shedding light on its potential as a systematic blueprint for #AGI ✨🚀
deniseholt.us/dr-karl-fristo…
#ActiveInferenceAI #KarlFriston #IntelligentAgents #VERSESAI #FreeEnergyPrinciple #AIS #FutureofAI #FirstPrinciplesAI

1
4
684
13 Nov 2023
NEW EPISODE: NOV 8th, I had a wonderfully engaging conversation, w/ global #AI expert, Dr. Jacques Ludik, re future of intelligent systems & how to realize AI’s full potential.
Watch here➡️ bit.ly/47xIIka
#ActiveInference #FirstPrinciplesAI #FEP #FutureofAI #HSML #AIGovernance #VERSES
8
9
259
7 Nov 2023
NEW EPISODE: On Oct 19, I had the great pleasure of interviewing Professor Bert De Vries, one of the world's leading researchers on #ActiveInference & #FirstPrinciplesAI
Check out the conversation here➡️youtu.be/vKhpkKvv2Tk?si=10uv…
#AI #FreeEnergyPrinciple #KarlFriston
5
730