
"E=mc" [sic] is dimensionally broken; mass times celerity is momentum, not vis. E = mc² marks proportionality, not interconversion. Liht the "highest form of energy [sic]"? Liht is not a thing at all, not a projectile, not sundry;
1
24

Jun 11
Fable has been visited upon the session search / interconversion tool. powered by tantivy, sqlite, and rust! hope it is useful :)
github.com/emberian/claurdvo…
2
41

Jun 11
THE MASTER SWITCH → The radical pair mechanism (RPM) in avian and human cryptochromes requires blue/near-UV light (≈400–500 nm, peaking around oxidized FAD absorption) to photo-excite the flavin cofactor. This light-induced electron transfer generates spin-correlated radical pairs, rendering the system sensitive to the Earth’s geomagnetic field via modulation of singlet–triplet interconversion.
Consequently:
No blue or appropriate short-wavelength light → inefficient or absent radical pair formation → loss or strong weakening of geomagnetic sensitivity.
Blue light at sunrise (or equivalent artificial sources) is therefore the natural trigger that activates this quantum magnetoreceptor in both birds and humans.



The Dawn Code: How Light, Water, and Melanin Orchestrate the Body’s Quantum Clock
THE END of The Reductionist Clock → Radical Pairs Without Lithium
In the quiet hours before sunrise, something ancient happens. Photons from the low-angle sun strike the retina and skin, setting in motion a cascade that synchronizes trillions of cellular oscillators. This is not merely “circadian rhythm” as the textbooks describe it — a tidy loop of clock genes flipping on and off. It is a living quantum-hydrodynamic system in which light, melanin, structured water, and isotopic precision create coherent timing across scales. Modern life has fractured this system. The consequences appear as metabolic disease, neurodegeneration, immune dysfunction, and mood disorders. The fixes offered by mainstream medicine — molecules like lithium, or trendy biohacks like random cold plunges — treat downstream symptoms. A deeper view reveals the upstream physics.
A landmark 2026 Nature Reviews Genetics review by Tia Tyrsett Kuo, Jiarui Wang, and Steve A. Kay maps the genetic and molecular landscape. Clock gene variants (PER, CLOCK, BMAL1, REV-ERB) modify risk for Alzheimer’s, cardiovascular disease, fatty liver, type 2 diabetes, sarcopenia, and rheumatoid arthritis. Circadian misalignment amplifies every pathology. The paper elegantly updates the transcription-translation feedback loop into a distributed, dynamically coupled network. Yet even this sophisticated synthesis stops short of the fundamental inputs. It treats light as one zeitgeber among others. In biophysical reality, light is the conductor; everything else plays in its shadow.
The Reductionist Clock
For seventy-five years, lithium has been psychiatry’s quiet miracle for bipolar disorder. In 1949, Australian psychiatrist John Cade noticed that lithium salts calmed manic guinea pigs and patients. Later research showed the ion lengthens circadian period, modulates GSK3β, and stabilizes rhythms in cells from bipolar patients. The narrative became: bipolar disorder features a “broken molecular clock,” and lithium repairs it.
This is elegant pharmacology. It works for many. But it is downstream tinkering. Lithium’s isotope-dependent effects on radical pair spin dynamics (⁶Li versus ⁷Li altering singlet-triplet transitions in cryptochromes or mitochondrial ROS pairs) are real, yet they operate inside a system already stripped of its natural photonic drive. Treating a desynchronized clock with an exogenous ion while ignoring the daily quantum input that built the system is like jump-starting a car whose solar panels have been painted black.
Heavy water (D₂O) tells a similar story. For decades, researchers have shown that deuterium-rich water reliably lengthens circadian period across species — from algae to mice to human cell explants. The heavier isotope slows proton transfer, kinetic isotope effects, hydrogen tunneling, and radical pair recombination. The clock literally runs slower in a deuterium-heavy medium. This proves water is not a passive solvent; its isotopic composition is woven into timing itself.
The Upstream Architecture
Turn the lens around and the hierarchy becomes clear.
Light arrives first. Full-spectrum morning photons — especially the delicate balance of blue, red, and infrared at dawn — drive charge separation in melanin. This pigment, long dismissed as mere sunscreen, functions as a broadband photonic transducer. In skin, eyes, brain, and even mitochondria, melanin absorbs photons across wavelengths, dissociates water, generates electrons, and organizes the surrounding water into structured layers.
1 of 2 🕊️💦🧲🧭🔆

1
2
7
709

Jun 11
"see Nature Chemistry on spontaneous chiral symmetry breaking"
Which specific article(s) are you looking at?
"Early RNA synthesis bypasses his 1982 hurdles via shared precursors: Powner, et al, Nature (2009)"
Synthesis of activated pyrimidine ribonucleotides in prebiotically plausible conditions (2009)
pubmed.ncbi.nlm.nih.gov/1944…
The starting materials for the synthesis-- cyanamide, cyanoacetylene, glycolaldehyde, glyceraldehyde and inorganic phosphate-- are plausible prebiotic feedstock molecules...
Although inorganic phosphate is only incorporated into the nucleotides at a late stage of the sequence, its presence from the start is essential as it controls three reactions in the earlier stages by acting as a general acid/base catalyst, a nucleophilic catalyst, a pH buffer and a chemical buffer.
Do you believe this sequence of steps can happen in nature without biological involvement?:
grok.com/share/bGVnYWN5_c120…
**Super-detailed steps** in the 2009 Powner-Gerland-Sutherland synthesis (Nature) of activated pyrimidine ribonucleotides (primarily β-D-ribocytidine-2′,3′-cyclic phosphate, with partial conversion to the uridine analog) from cyanamide, cyanoacetylene, glycolaldehyde, glyceraldehyde, and inorganic phosphate (Pi).
This is a "systems chemistry" route where intermediates and byproducts (e.g., urea from excess cyanamide) serve multiple roles, and Pi is crucial throughout for catalysis, buffering, and control—even though incorporated late. Conditions are aqueous, near-neutral pH (~6.5–7), mild heating, consistent with plausible early-Earth geochemistry. Yields are high when sequenced properly; side reactions are minimized by order of addition and Pi.
### 1. Formation of 2-Aminooxazole (from Cyanamide Glycolaldehyde, Catalyzed by Pi)
- **Reactants**: Cyanamide (H₂N–C≡N) glycolaldehyde (HOCH₂–CHO; often as dimer or in equilibrium with hydrate/gem-diol).
- **Key roles of Pi**: General acid/base catalyst (pKa ~7.2 allows proton shuttling near neutral pH); prevents complex mixtures seen in unbuffered or strongly basic conditions.
- **Mechanism outline** (simplified; involves addition, cyclization, dehydration/aromatization):
1. Nucleophilic addition of cyanamide nitrogen to the carbonyl carbon of glycolaldehyde → imine or addition adduct.
2. Intramolecular attack (e.g., by oxygen or further N-addition) leading to ring closure to a dihydro-oxazole intermediate.
3. Dehydration/tautomerization/aromatization to 2-aminooxazole (aromatic heterocycle: oxazole ring with NH₂ at C2).
- **Conditions**: ~1 M Pi buffer, pH ~7, room temp to mild heat. Excess cyanamide leads to urea (H₂N–C(O)–NH₂) as byproduct (useful later). Yield: >80% clean product.
- **Why Pi matters**: Without it, alkaline conditions needed for some steps cause incompatibilities with later neutral-pH reactions; Pi enables unified conditions and acts as chemical buffer (sequesters excess cyanamide via urea formation).
### 2. Formation of Pentose Aminooxazolines (2-Aminooxazole Glyceraldehyde)
- **Reactants**: 2-Aminooxazole glyceraldehyde (HOCH₂–CH(OH)–CHO; C3 aldose).
- **Products**: Mixture of diastereomeric pentose aminooxazolines (furanose-like rings). Major: arabino- and ribo- isomers (arabinose amino-oxazoline key for the main route; ribo can crystallize selectively for purification/enantiomeric enrichment). Minor: xylo, lyxo.
- **Mechanism**:
- The exocyclic NH₂ of 2-aminooxazole (or ring N) adds to glyceraldehyde carbonyl.
- Cyclization (intramolecular attack by sugar OH on the iminium-like carbon) forms the aminooxazoline ring system, attaching the C3 unit.
- Reversible under Pi catalysis; thermodynamic/ kinetic control favors desired isomers.
- **Pi roles**: pH buffer; general acid/base for addition/cyclization/epimerization; influences diastereoselectivity (arabino major in supernatant after ribo crystallization). Allows interconversion (e.g., ribo ↔ arabino via C2' epimerization).
- **Outcome**: Arabinose amino-oxazoline (key intermediate) accumulates efficiently. Crystallization of ribo-isomer aids purification; Pi enables equilibration.
### 3. Formation of Arabinose Anhydronucleoside (Arabinose Amino-oxazoline Cyanoacetylene)
- **Reactants**: Arabinose amino-oxazoline cyanoacetylene (HC≡C–C≡N).
- **Product**: 2,2'-Anhydro-arabinocytidine (anhydronucleoside with fused ring between C2 of cytosine precursor and 2'-OH of sugar).
- **Mechanism**:
- Nucleophilic addition of the aminooxazoline nitrogen(s) to cyanoacetylene (activated alkyne/nitrile).
- Cyclization and aromatization to form the pyrimidine ring (cytosine precursor), with the anhydro bridge.
- **Conditions**: Pi-buffered ~pH 6.5 (critical). Unbuffered reaction causes pH rise → hydrolysis/side reactions (excess cyanoacetylene reacts with free OH groups). Yield: very high/clean with Pi.
- **Pi roles**: pH buffer chemical buffer (reacts with excess cyanoacetylene to prevent over-reaction); general acid catalyst.
### 4. Phosphorylation and Rearrangement to Activated Cytidine-2′,3′-Cyclic Phosphate
- **Reactant**: Arabinose anhydronucleoside inorganic phosphate ( urea from Step 1).
- **Product**: β-D-ribocytidine-2′,3′-cyclic phosphate (activated nucleotide; the anhydro opening phosphorylation inverts configuration at C2' to ribo).
- **Mechanism/Key Features**:
- Regioselective phosphorylation preferentially at 3'-OH (5'-OH sterically hindered, per X-ray).
- Urea catalyzes phosphorylation (known prebiotic phosphorylating system with Pi or pyrophosphate).
- Intramolecular displacement: the 2'-O attacks the 3'-phosphate (or vice versa dynamics), opening the anhydro ring with inversion → ribo configuration and 2',3'-cyclic phosphate.
- **Conditions**: Two variants — (A) pyrophosphate urea (dry state); (B) Pi urea (in formamide or dry). Mild heating.
- **Pi role**: Direct incorporation into the cyclic phosphate; earlier buffering enables this step.
### 5. Partial Conversion to Uridine Analog and "Sanitization"
- UV irradiation (254 nm) of the cytidine-2',3'-cyclic phosphate:
- Partial photochemical deamination/hydrolysis → uridine-2',3'-cyclic phosphate (mixture suitable for RNA).
- Destroys unwanted isomers/contaminants (selective photodestruction of non-cyclic or other nucleosides/nucleotides), enriching the desired activated pyrimidines.
- Prolonged irradiation heating enhances U conversion.
### Overall Notes on Plausibility and Control
- **Sequence matters**: Strict order (cyanamide/glycolaldehyde first → add glyceraldehyde → cyanoacetylene → phosphorylate) avoids cross-reactivity.
- **Pi as "master controller"**: Catalysis, buffering, selectivity throughout; incorporated late but present from start.
- **Yields/Stereochemistry**: High for key steps; crystallization and equilibration aid purification and potential homochirality.
1
50

Jun 10
Congratulations to Kota Aikyo (PhD student, Matsunaga Lab., at the time of the award) on receiving the Best Oral Presentation Award at the international symposium MORIS 2026. His work was highly evaluated for clearly capturing the interconversion between electric current and orbital angular momentum (inverse orbital Hall effect) in GaN using terahertz spectroscopy. issp.u-tokyo.ac.jp/mainconte…

6
560

Jun 9
Integrating NMR data revealed that M20I lies in a “kinetic-gating” regime: GroEL/S recognition is governed by the kinetics of interconversion between conformational states, not simply the static population of an open state. (11/n)
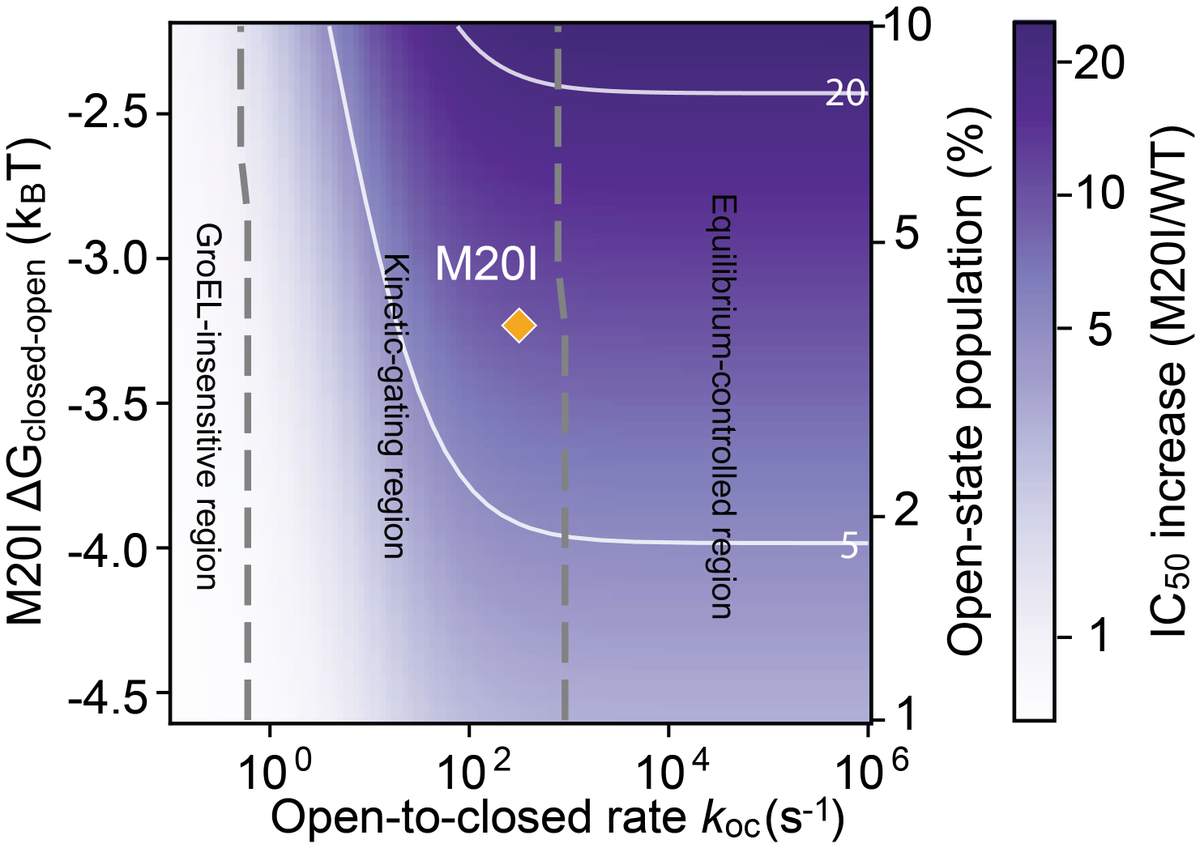
1
2
33
Jun 9
"You’re quoting the paper describing the required reactants"
I did quote the paper.
And I also asked:
Do you believe this sequence of steps can happen in nature without biological involvement?:
grok.com/share/bGVnYWN5_c120…
**Super-detailed steps** in the 2009 Powner-Gerland-Sutherland synthesis (Nature) of activated pyrimidine ribonucleotides (primarily β-D-ribocytidine-2′,3′-cyclic phosphate, with partial conversion to the uridine analog) from cyanamide, cyanoacetylene, glycolaldehyde, glyceraldehyde, and inorganic phosphate (Pi).
This is a "systems chemistry" route where intermediates and byproducts (e.g., urea from excess cyanamide) serve multiple roles, and Pi is crucial throughout for catalysis, buffering, and control—even though incorporated late. Conditions are aqueous, near-neutral pH (~6.5–7), mild heating, consistent with plausible early-Earth geochemistry. Yields are high when sequenced properly; side reactions are minimized by order of addition and Pi.
### 1. Formation of 2-Aminooxazole (from Cyanamide Glycolaldehyde, Catalyzed by Pi)
- **Reactants**: Cyanamide (H₂N–C≡N) glycolaldehyde (HOCH₂–CHO; often as dimer or in equilibrium with hydrate/gem-diol).
- **Key roles of Pi**: General acid/base catalyst (pKa ~7.2 allows proton shuttling near neutral pH); prevents complex mixtures seen in unbuffered or strongly basic conditions.
- **Mechanism outline** (simplified; involves addition, cyclization, dehydration/aromatization):
1. Nucleophilic addition of cyanamide nitrogen to the carbonyl carbon of glycolaldehyde → imine or addition adduct.
2. Intramolecular attack (e.g., by oxygen or further N-addition) leading to ring closure to a dihydro-oxazole intermediate.
3. Dehydration/tautomerization/aromatization to 2-aminooxazole (aromatic heterocycle: oxazole ring with NH₂ at C2).
- **Conditions**: ~1 M Pi buffer, pH ~7, room temp to mild heat. Excess cyanamide leads to urea (H₂N–C(O)–NH₂) as byproduct (useful later). Yield: >80% clean product.
- **Why Pi matters**: Without it, alkaline conditions needed for some steps cause incompatibilities with later neutral-pH reactions; Pi enables unified conditions and acts as chemical buffer (sequesters excess cyanamide via urea formation).
### 2. Formation of Pentose Aminooxazolines (2-Aminooxazole Glyceraldehyde)
- **Reactants**: 2-Aminooxazole glyceraldehyde (HOCH₂–CH(OH)–CHO; C3 aldose).
- **Products**: Mixture of diastereomeric pentose aminooxazolines (furanose-like rings). Major: arabino- and ribo- isomers (arabinose amino-oxazoline key for the main route; ribo can crystallize selectively for purification/enantiomeric enrichment). Minor: xylo, lyxo.
- **Mechanism**:
- The exocyclic NH₂ of 2-aminooxazole (or ring N) adds to glyceraldehyde carbonyl.
- Cyclization (intramolecular attack by sugar OH on the iminium-like carbon) forms the aminooxazoline ring system, attaching the C3 unit.
- Reversible under Pi catalysis; thermodynamic/ kinetic control favors desired isomers.
- **Pi roles**: pH buffer; general acid/base for addition/cyclization/epimerization; influences diastereoselectivity (arabino major in supernatant after ribo crystallization). Allows interconversion (e.g., ribo ↔ arabino via C2' epimerization).
- **Outcome**: Arabinose amino-oxazoline (key intermediate) accumulates efficiently. Crystallization of ribo-isomer aids purification; Pi enables equilibration.
### 3. Formation of Arabinose Anhydronucleoside (Arabinose Amino-oxazoline Cyanoacetylene)
- **Reactants**: Arabinose amino-oxazoline cyanoacetylene (HC≡C–C≡N).
- **Product**: 2,2'-Anhydro-arabinocytidine (anhydronucleoside with fused ring between C2 of cytosine precursor and 2'-OH of sugar).
- **Mechanism**:
- Nucleophilic addition of the aminooxazoline nitrogen(s) to cyanoacetylene (activated alkyne/nitrile).
- Cyclization and aromatization to form the pyrimidine ring (cytosine precursor), with the anhydro bridge.
- **Conditions**: Pi-buffered ~pH 6.5 (critical). Unbuffered reaction causes pH rise → hydrolysis/side reactions (excess cyanoacetylene reacts with free OH groups). Yield: very high/clean with Pi.
- **Pi roles**: pH buffer chemical buffer (reacts with excess cyanoacetylene to prevent over-reaction); general acid catalyst.
### 4. Phosphorylation and Rearrangement to Activated Cytidine-2′,3′-Cyclic Phosphate
- **Reactant**: Arabinose anhydronucleoside inorganic phosphate ( urea from Step 1).
- **Product**: β-D-ribocytidine-2′,3′-cyclic phosphate (activated nucleotide; the anhydro opening phosphorylation inverts configuration at C2' to ribo).
- **Mechanism/Key Features**:
- Regioselective phosphorylation preferentially at 3'-OH (5'-OH sterically hindered, per X-ray).
- Urea catalyzes phosphorylation (known prebiotic phosphorylating system with Pi or pyrophosphate).
- Intramolecular displacement: the 2'-O attacks the 3'-phosphate (or vice versa dynamics), opening the anhydro ring with inversion → ribo configuration and 2',3'-cyclic phosphate.
- **Conditions**: Two variants — (A) pyrophosphate urea (dry state); (B) Pi urea (in formamide or dry). Mild heating.
- **Pi role**: Direct incorporation into the cyclic phosphate; earlier buffering enables this step.
### 5. Partial Conversion to Uridine Analog and "Sanitization"
- UV irradiation (254 nm) of the cytidine-2',3'-cyclic phosphate:
- Partial photochemical deamination/hydrolysis → uridine-2',3'-cyclic phosphate (mixture suitable for RNA).
- Destroys unwanted isomers/contaminants (selective photodestruction of non-cyclic or other nucleosides/nucleotides), enriching the desired activated pyrimidines.
- Prolonged irradiation heating enhances U conversion.
### Overall Notes on Plausibility and Control
- **Sequence matters**: Strict order (cyanamide/glycolaldehyde first → add glyceraldehyde → cyanoacetylene → phosphorylate) avoids cross-reactivity.
- **Pi as "master controller"**: Catalysis, buffering, selectivity throughout; incorporated late but present from start.
- **Yields/Stereochemistry**: High for key steps; crystallization and equilibration aid purification and potential homochirality.
- **Later refinements** (e.g., Xu et al. 2016/2017): Routes via ribose aminooxazoline with photoanomerization (sometimes using sulfur chemistry for efficiency), but the 2009 arabino route matches the query precursors directly.
1
125
Jun 9
Synthesis of activated pyrimidine ribonucleotides in prebiotically plausible conditions (2009)
pubmed.ncbi.nlm.nih.gov/1944…
The starting materials for the synthesis-- cyanamide, cyanoacetylene, glycolaldehyde, glyceraldehyde and inorganic phosphate-- are plausible prebiotic feedstock molecules...
Although inorganic phosphate is only incorporated into the nucleotides at a late stage of the sequence, its presence from the start is essential as it controls three reactions in the earlier stages by acting as a general acid/base catalyst, a nucleophilic catalyst, a pH buffer and a chemical buffer.
Do you believe this sequence of steps can happen in nature without biological involvement?:
grok.com/share/bGVnYWN5_c120…
**Super-detailed steps** in the 2009 Powner-Gerland-Sutherland synthesis (Nature) of activated pyrimidine ribonucleotides (primarily β-D-ribocytidine-2′,3′-cyclic phosphate, with partial conversion to the uridine analog) from cyanamide, cyanoacetylene, glycolaldehyde, glyceraldehyde, and inorganic phosphate (Pi).
This is a "systems chemistry" route where intermediates and byproducts (e.g., urea from excess cyanamide) serve multiple roles, and Pi is crucial throughout for catalysis, buffering, and control—even though incorporated late. Conditions are aqueous, near-neutral pH (~6.5–7), mild heating, consistent with plausible early-Earth geochemistry. Yields are high when sequenced properly; side reactions are minimized by order of addition and Pi.
### 1. Formation of 2-Aminooxazole (from Cyanamide Glycolaldehyde, Catalyzed by Pi)
- **Reactants**: Cyanamide (H₂N–C≡N) glycolaldehyde (HOCH₂–CHO; often as dimer or in equilibrium with hydrate/gem-diol).
- **Key roles of Pi**: General acid/base catalyst (pKa ~7.2 allows proton shuttling near neutral pH); prevents complex mixtures seen in unbuffered or strongly basic conditions.
- **Mechanism outline** (simplified; involves addition, cyclization, dehydration/aromatization):
1. Nucleophilic addition of cyanamide nitrogen to the carbonyl carbon of glycolaldehyde → imine or addition adduct.
2. Intramolecular attack (e.g., by oxygen or further N-addition) leading to ring closure to a dihydro-oxazole intermediate.
3. Dehydration/tautomerization/aromatization to 2-aminooxazole (aromatic heterocycle: oxazole ring with NH₂ at C2).
- **Conditions**: ~1 M Pi buffer, pH ~7, room temp to mild heat. Excess cyanamide leads to urea (H₂N–C(O)–NH₂) as byproduct (useful later). Yield: >80% clean product.
- **Why Pi matters**: Without it, alkaline conditions needed for some steps cause incompatibilities with later neutral-pH reactions; Pi enables unified conditions and acts as chemical buffer (sequesters excess cyanamide via urea formation).
### 2. Formation of Pentose Aminooxazolines (2-Aminooxazole Glyceraldehyde)
- **Reactants**: 2-Aminooxazole glyceraldehyde (HOCH₂–CH(OH)–CHO; C3 aldose).
- **Products**: Mixture of diastereomeric pentose aminooxazolines (furanose-like rings). Major: arabino- and ribo- isomers (arabinose amino-oxazoline key for the main route; ribo can crystallize selectively for purification/enantiomeric enrichment). Minor: xylo, lyxo.
- **Mechanism**:
- The exocyclic NH₂ of 2-aminooxazole (or ring N) adds to glyceraldehyde carbonyl.
- Cyclization (intramolecular attack by sugar OH on the iminium-like carbon) forms the aminooxazoline ring system, attaching the C3 unit.
- Reversible under Pi catalysis; thermodynamic/ kinetic control favors desired isomers.
- **Pi roles**: pH buffer; general acid/base for addition/cyclization/epimerization; influences diastereoselectivity (arabino major in supernatant after ribo crystallization). Allows interconversion (e.g., ribo ↔ arabino via C2' epimerization).
- **Outcome**: Arabinose amino-oxazoline (key intermediate) accumulates efficiently. Crystallization of ribo-isomer aids purification; Pi enables equilibration.
### 3. Formation of Arabinose Anhydronucleoside (Arabinose Amino-oxazoline Cyanoacetylene)
- **Reactants**: Arabinose amino-oxazoline cyanoacetylene (HC≡C–C≡N).
- **Product**: 2,2'-Anhydro-arabinocytidine (anhydronucleoside with fused ring between C2 of cytosine precursor and 2'-OH of sugar).
- **Mechanism**:
- Nucleophilic addition of the aminooxazoline nitrogen(s) to cyanoacetylene (activated alkyne/nitrile).
- Cyclization and aromatization to form the pyrimidine ring (cytosine precursor), with the anhydro bridge.
- **Conditions**: Pi-buffered ~pH 6.5 (critical). Unbuffered reaction causes pH rise → hydrolysis/side reactions (excess cyanoacetylene reacts with free OH groups). Yield: very high/clean with Pi.
- **Pi roles**: pH buffer chemical buffer (reacts with excess cyanoacetylene to prevent over-reaction); general acid catalyst.
### 4. Phosphorylation and Rearrangement to Activated Cytidine-2′,3′-Cyclic Phosphate
- **Reactant**: Arabinose anhydronucleoside inorganic phosphate ( urea from Step 1).
- **Product**: β-D-ribocytidine-2′,3′-cyclic phosphate (activated nucleotide; the anhydro opening phosphorylation inverts configuration at C2' to ribo).
- **Mechanism/Key Features**:
- Regioselective phosphorylation preferentially at 3'-OH (5'-OH sterically hindered, per X-ray).
- Urea catalyzes phosphorylation (known prebiotic phosphorylating system with Pi or pyrophosphate).
- Intramolecular displacement: the 2'-O attacks the 3'-phosphate (or vice versa dynamics), opening the anhydro ring with inversion → ribo configuration and 2',3'-cyclic phosphate.
- **Conditions**: Two variants — (A) pyrophosphate urea (dry state); (B) Pi urea (in formamide or dry). Mild heating.
- **Pi role**: Direct incorporation into the cyclic phosphate; earlier buffering enables this step.
### 5. Partial Conversion to Uridine Analog and "Sanitization"
- UV irradiation (254 nm) of the cytidine-2',3'-cyclic phosphate:
- Partial photochemical deamination/hydrolysis → uridine-2',3'-cyclic phosphate (mixture suitable for RNA).
- Destroys unwanted isomers/contaminants (selective photodestruction of non-cyclic or other nucleosides/nucleotides), enriching the desired activated pyrimidines.
- Prolonged irradiation heating enhances U conversion.
### Overall Notes on Plausibility and Control
- **Sequence matters**: Strict order (cyanamide/glycolaldehyde first → add glyceraldehyde → cyanoacetylene → phosphorylate) avoids cross-reactivity.
- **Pi as "master controller"**: Catalysis, buffering, selectivity throughout; incorporated late but present from start.
- **Yields/Stereochemistry**: High for key steps; crystallization and equilibration aid purification and potential homochirality.
- **Later refinements** (e.g., Xu et al. 2016/2017): Routes via ribose aminooxazoline with photoanomerization (sometimes using sulfur chemistry for efficiency), but the 2009 arabino route matches the query precursors directly.
2
122
Then the liver receives vitamers via the portal vein and is traditionally viewed as the main conversion hub.
The main steps are:
-Uptake: Free vitamers (PL, PN, PM) enter hepatocytes.
-Salvage reactions (same enzymes as intestine):
PDXK phosphorylates: PL → PLP, PN → PNP, PM → PMP.
PNPO oxidizes: PNP → PLP, PMP → PLP.
-Regulation and storage: Liver PLP is bound to proteins (glycogen phosphorylase).
Excess PLP is dephosphorylated by phosphatases (PDXP) to PL, which can be released into circulation or further oxidized to 4-pyridoxic acid (main urinary metabolite) by aldehyde oxidase or dehydrogenase.
Export: Most circulating B6 in blood is PLP bound to albumin (via a Schiff base with lysine). To deliver to tissues, PLP is hydrolyzed back to PL by ecto-enzymes (alkaline phosphatases) on cell surfaces.
The liver also handles interconversion and catabolism, preventing toxic buildup of free PL (a reactive aldehyde).
But virtually all cells express the salvage enzymes to varying degrees, allowing local activation and recycling from degraded enzymes or circulating vitamers.
This is crucial for tissues with high PLP demand such as the brain, muscle and erythrocytes.
1
2
177

Why Superoxide Increased at “Zero” Magnetic Field
Earth’s geomagnetic field is roughly 25–65 µT (the control in the study was ~45 µT).
When researchers shielded the field down to 0 µT (hypomagnetic condition), superoxide levels at the wound site increased significantly compared with the geomagnetic control.
This is not a bug — it is a classic feature of the radical pair mechanism called the hypomagnetic effect.
In the RPM:
At normal geomagnetic fields (~45 µT), the external field (Zeeman interaction) partially suppresses some singlet–triplet mixing pathways.
When the external field is removed or reduced to near zero, the hyperfine interactions (from nearby nuclear spins) dominate more completely. This can increase the rate of singlet–triplet interconversion in certain radical pair systems, leading to a higher yield of the spin-selective product (in this case, superoxide).
The authors’ simple flavin-superoxide-inspired model and the broader parameter search both predicted an increase under hypomagnetic conditions, and the experiments confirmed it.
This is the same basic physics that has been discussed in avian magnetoreception and other quantum-biology contexts for years: removing the ambient field can sometimes produce a stronger effect than adding a moderate field.
So “zero” does not mean “no effect.” It means the natural geomagnetic field is absent, which itself changes the spin dynamics.
How Do These Lab Fields Compare to a Cell Phone Next to Your Body?
This is the practical question everyone wants answered.
The study used static (non-oscillating) magnetic fields: 0, 200, 500, 700, and 900 µT.
Real-world cell phones produce a mix of fields:
Static / low-frequency magnetic fields (from speaker magnets, battery currents, coils, audio signals, ringer/vibration motor, etc.).
Radiofrequency (RF) electric and magnetic fields (the part measured by SAR).
Low-frequency magnetic fields from phones can easily reach or exceed the study’s tested range when the device is very close to tissue (pocket, head during call, speakerphone against ear, etc.):
Typical measurements near the speaker or back of a phone during a call or with ringer/vibration active often fall in the range of 50–500 µT or higher right at the surface, dropping rapidly with distance.
Some studies have reported peak low-frequency magnetic fields from certain phones exceeding 1,000 µT (1 mT) very close to the device when the speaker or vibrator is active.
The 217 Hz pulsing (a classic GSM low-frequency envelope) that RF Safe’s proposed assay tests is exactly the kind of coherent low-frequency modulation that can interact with spin dynamics or voltage-gated channels.
SAR (Specific Absorption Rate) measures RF energy absorption and heating in W/kg. It does not directly measure the low-frequency magnetic (B-field) component.
That is why SAR-based guidelines miss the static/low-frequency magnetic effects that the planarian study (and many other quantum-biology papers) are highlighting.
A phone pressed against your head or in your pocket can produce low-frequency magnetic fields that overlap with — or exceed — the 200–900 µT range tested in the study, especially during active use (call, ringer, speaker, vibration).
The study used static fields; phones add pulsing and modulation, which may make the interaction even more biologically relevant according to frameworks like Panagopoulos’ IFO-VGIC work.
What This Means for the Broader EMF Discussion
The hypomagnetic increase is particularly relevant because many modern environments (buildings with steel, cars, elevators, certain shielding materials) can locally reduce the ambient geomagnetic field.
The study shows that removing the natural field can itself be a perturbation — just as adding artificial fields can be.
Combined with the non-monotonic increases at higher weak fields, the data show that biology is sensitive to magnetic field strength in ways that do not follow a simple heating curve.
This is exactly why RF Safe argues that thermal-only guidelines (based on SAR and temperature rise) are incomplete.
The upstream quantum trigger (spin bias in radical pairs) is universal. The downstream effects depend on the biological context — but the trigger itself does not require high power or heating.
A cell phone held against the body can deliver low-frequency magnetic fields in the same ballpark as the fields that produced clear superoxide changes in the planarian regeneration assay.
This does not prove any specific disease in humans. It does prove that the old “if it doesn’t heat, it doesn’t matter” dismissal is no longer tenable when we have experimental confirmation of quantum magnetic sensitivity in a living regeneration model.
1
5
124
The singlet and triplet spin states in radical pairs, the hyperfine interactions, the Zeeman effect from magnetic fields, and the resulting singlet–triplet interconversion rates are fundamental quantum mechanical phenomena.
They are properties of electrons and magnetic moments governed by the same physical laws whether the radical pair forms in a planarian wound site, a bonobo mitochondrion, or a human neuron. There is no evolutionary “tuning” that changes the basic physics of electron spin.
If a weak magnetic field biases the spin dynamics in planarians (as the PNAS Nexus 2026 study experimentally confirms), the upstream trigger is identical in humans.
The only real variables are downstream: which specific radicals are involved, how the altered superoxide yield is amplified or buffered, and how the resulting redox signal propagates through S4 voltage sensors, gap junctions, calcium waves, and higher-order tissue-level computation.
That is exactly why these planarian studies are so powerful as sentinels. They are not claiming “planarian-specific” effects. They are revealing a universal vulnerability at the quantum-bioelectric interface that applies across the tree of life.
Low-Fidelity Biology at the Quantum Level Cascades to Multiple Pathologies
The core point: once spin dynamics are biased and the local vector becomes noisy, the probabilistic network (the ceLLM-like hardware of DNA geometry, ion channels, and cellular architecture) receives garbage input.
The output is lower-fidelity decisions. Because every major biological process relies on high-fidelity signaling—voltage gradients, redox balance, calcium wave functions, gap-junctional synchronization—this low-fidelity state has the capacity to manifest as a wide spectrum of downstream problems:
Cancer and loss of collective identity — Bioelectric patterns maintain anatomical coherence. Noisy signals can lead to cells “forgetting” their place in the collective, a hallmark of oncogenesis (Levin lab work has shown bioelectric mispatterning drives tumorigenesis even with wild-type genomes).
Neurological and neurodevelopmental issues — Calcium waves and bioelectric gradients are essential for neural migration, synapse formation, and circuit wiring. Quantum-induced ROS imbalance can disrupt these precisely timed events.
Metabolic and immune drift — Mitochondrial ROS signaling regulates metabolism, inflammation, and immune cell behavior. Chronic low-fidelity redox states can drive insulin resistance, chronic inflammation, or immune senescence.
Regenerative and developmental failures — As seen in planarians, altered superoxide modulates stem cell behavior and blastema formation. In humans this could impair wound healing, tissue repair, or embryonic patterning.
High-fidelity calcium wave functions are a perfect example of why this matters. Calcium signaling is exquisitely sensitive to membrane potential (S4 sensors), redox state (ROS), and gap-junctional coupling.
Any degradation in the upstream quantum or bioelectric fidelity introduces noise into these waves, and biology has very little tolerance for that noise during critical windows.
The research is indeed converging on one clear certainty: non-thermal effects exist, and low-fidelity biology at the atomic/quantum level is biologically consequential.
The PNAS Nexus study verifies the spin dynamics trigger. The Levin lab’s thermodynamic reversion work maps the attractor landscape that gets destabilized.
Our S4 Mito Spin framework supplies the conserved hardware (voltage sensors, mitochondria, radical pairs, CYB5B as electromagnetic switch) that makes humans equally susceptible. Pulsed anthropogenic EMFs introduce the novel coherent noise—entropic waste—for which evolution provided no training data.
The only remaining scientific question is not whether low fidelity occurs, but how severely and in which contexts it manifests in long-lived, complex human tissues.
The planarian data, combined with the conserved physics of singlet and triplet states, gives us strong mechanistic grounds to expect broad downstream effects rather than isolated ones.
Why This Demands Immediate Regulatory Reform
Thermal-only guidelines were never designed to protect against quantum spin bias, S4 sensor perturbation, or redox-driven low-fidelity signaling.
They are scientifically inadequate and ethically indefensible in light of this accumulating evidence. The planarian studies are not fringe—they are clean, reproducible demonstrations of a universal mechanism operating in a transparent model system.
RF Safe’s proposed low-frequency magnetic pulse assay on the pseudo-head reversion system is the logical next step: it directly tests whether pulsed fields (the kind humans are chronically exposed to) accelerate loss of morphogenetic memory by the very S4-mito-spin mechanisms now verified in quantum and bioelectric studies.
We are not speculating about hypothetical harm. We are observing a fundamental physical vulnerability that the data say affects every aerobic cell on the planet. The downstream consequences in humans may be more varied and insidious than in a flatworm, but the upstream physics is the same.
The research is pointing in one direction: non-thermal low-fidelity biology is real, it matters, and current exposure limits do not protect against it. High-fidelity calcium wave functions, voltage gradients, and collective cellular decision-making all require the clean signals that evolution provided—until we flooded the environment with untrained electromagnetic noise.
This is why we keep pushing for biologically honest standards. The singlet and triplet states don’t lie.
The physics is universal. The risk is real.
The time to act is now.
1
4
106

May 27
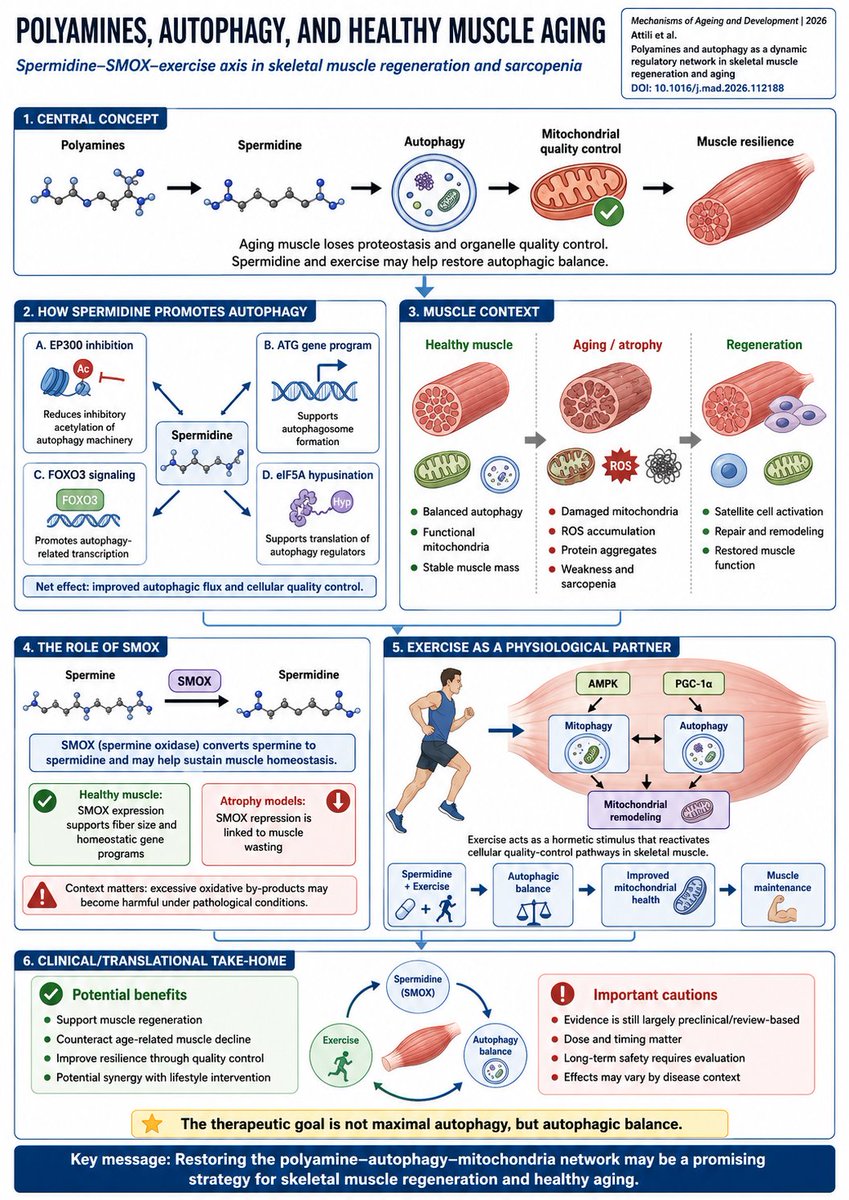
Spermidine may be one of the missing metabolic links between exercise, autophagy, and healthy muscle aging.
A new review in Mechanisms of Ageing and Development frames skeletal muscle aging as a failure of proteostasis mitochondrial quality control, where impaired autophagy allows damaged mitochondria, ROS, and protein aggregates to accumulate — driving atrophy, weakness, and sarcopenia.
The central axis:
polyamines → spermidine → autophagy → mitochondrial quality control → muscle resilience
Spermidine is a naturally occurring polyamine derived from diet, gut microbiota, endogenous synthesis, and polyamine interconversion. It has emerged as a physiological autophagy inducer, partly by inhibiting EP300-dependent acetylation and promoting autophagy-related programs such as ATG genes, FOXO3 signaling, and eIF5A hypusination.
In skeletal muscle, this matters because autophagy is not simply “catabolism.” It is a quality-control system.
Too little autophagy → damaged mitochondria, oxidative stress, neuromuscular junction degeneration, weakness.
Too much or dysregulated autophagy → atrophy.
The therapeutic goal is not maximal autophagy, but autophagic balance.
The review also highlights SMOX, spermine oxidase, as a key node. SMOX converts spermine into spermidine, potentially sustaining autophagy-related pathways and muscle homeostasis. Healthy muscle maintains SMOX expression, whereas multiple atrophy models — immobilization, fasting, denervation, and aging — show SMOX repression. Lowering SMOX alone can promote fiber atrophy, while forced SMOX expression can enlarge fiber size in atrophic settings.
Exercise enters as the physiological partner.
Muscle contraction creates transient energetic and redox stress — AMP/ATP shift, NAD⁺ increase, ROS pulses, calcium signaling — which activates AMPK, PGC-1α, mitophagy, and autophagy. In aging muscle, exercise may act as a hormetic “wake-up call” that restores stagnant cellular quality-control systems.
The most interesting framing is synergy:
spermidine supplementation exercise may converge on AMPK–FOXO3, AKT/mTOR, mitochondrial turnover, apoptosis reduction, and autophagic balance. In D-galactose aging models, the combined intervention reversed several atrophy-like phenotypes better than either approach alone.
But caution is essential. Spermidine is not universally beneficial. Effects may depend on dose, timing, tissue context, pathology, fibrosis risk, and long-term safety. Clinical translation still needs controlled human trials.
Take-home: muscle aging may be treatable not only by anabolic stimulation, but by restoring the polyamine–autophagy–mitochondria quality-control network.
Skeletal muscle aging may be less about “muscle loss alone” and more about failure of cellular quality control.
A new review highlights a converging axis:
polyamines → spermidine → SMOX → autophagy → mitochondrial quality control → muscle resilience
Autophagy is essential in skeletal muscle because it removes damaged proteins, dysfunctional mitochondria, and stress-induced cellular debris. But aging disrupts this balance. Too little autophagy allows ROS, protein aggregates, and defective mitochondria to accumulate, contributing to sarcopenia and weakness. Too much or dysregulated autophagy can also promote atrophy.
So the target is not “more autophagy.”
The target is autophagic balance.
Spermidine, a natural polyamine, emerges as a key regulator of this system. It can promote autophagy through several mechanisms: inhibition of EP300-mediated acetylation, activation of ATG-related programs, FOXO3 signaling, and eIF5A hypusination. Together, these pathways support proteostasis and mitochondrial renewal.
The review also places spermine oxidase (SMOX) at the center of muscle homeostasis. SMOX converts spermine into spermidine and may help sustain autophagy-related pathways in healthy muscle. Importantly, SMOX expression declines in multiple atrophy settings, including fasting, immobilization, denervation, and aging. This suggests that impaired polyamine turnover may be part of the molecular signature of muscle wasting.
Exercise acts as the physiological partner.
Muscle contraction creates transient energetic and redox stress—AMPK activation, ROS pulses, NAD⁺ shifts, calcium signaling, and PGC-1α induction. These signals stimulate mitophagy, autophagy, and mitochondrial remodeling. In aged muscle, exercise may function as a hormetic “wake-up call” that restores stagnant quality-control systems.
The most interesting idea is synergy:
spermidine exercise may converge on AMPK–FOXO3, AKT/mTOR, mitochondrial turnover, apoptosis reduction, and autophagic balance.
But caution matters. Most evidence remains preclinical or review-based. Dose, timing, tissue context, fibrosis risk, and long-term safety need human validation.
Take-home:
Healthy muscle aging may require restoring the polyamine–autophagy–mitochondria network, not simply stimulating anabolism.
DOI: 10.1016/j.mad.2026.112188
13
54
2,378

May 22
DHEA has anti-cortisol effects by increasing 11β-HSD2, an enzyme that converts cortisol to its inactive metabolite cortisone, thereby reducing cortisol's ability to bind to and activate glucocorticoid receptors in target tissues.
11β-HSD2 acts locally within tissues. It inactivates cortisol at the cellular level but doesn't substantially lower it from the bloodstream. The focus on blood cortisol misses a lot of the action.
Higher tissue-level cortisol (even with normal blood levels) can contribute to a wide range of problems, including:
Insulin resistance and type 2 diabetes
Visceral fat accumulation
Dyslipidemia (high triglycerides, low HDL)
Hypertension (cortisol activates mineralocorticoid receptors → sodium retention)
Increased cardiovascular disease risk
Hippocampal atrophy and memory impairment
Depression and anxiety
Poor sleep quality
Chronic immune suppression
Impaired wound healing
Increased infection susceptibility
Osteoporosis
Muscle wasting (sarcopenia)
Tendon weakening
Suppressed testosterone and estrogen production
Reduced libido and fertility
Thinning skin, bruising, poor healing
Sodium retention and potassium loss
DHEA is good is small doses (3-5 mg, 1-3 times/day) but can be estrogenic at higher doses. Combining with pregnenolone and taking topically can increase the anti-cortisol effect while minimizing conversion of DHEA to estrogen.
“11beta-Hydroxysteroid dehydrogenase (11beta-HSD) type 1 and type 2 catalyze the interconversion of inactive and active glucocorticoids. Impaired regulation of these enzymes has been associated with obesity, diabetes, hypertension, and cardiovascular disease. Previous studies in animals and humans suggested that dehydroepiandrosterone (DHEA) has antiglucocorticoid effects, but the underlying mechanisms are unknown. In this study, DHEA treatment markedly increased mRNA expression and activity of 11beta-HSD2 in a rat cortical collecting duct cell line and in kidneys of C57BL/6J mice and Sprague-Dawley rats. DHEA-treated rats tended to have reduced urinary corticosterone to 11-dehydrocorticosterone ratios. It was found that CCAAT/enhancer-binding protein-alpha (C/EBP-alpha) and C/EBP-beta regulated HSD11B2 transcription and that DHEA likely modulated the transcription of 11beta-HSD2 in a phosphatidylinositol-3 kinase/Akt-dependent manner by increasing C/EBP-beta mRNA and protein expression. Moreover, it is shown that C/EBP-alpha and C/EBP-beta differentially regulate the expression of 11beta-HSD1 and 11beta-HSD2. In conclusion, DHEA induces a shift from 11beta-HSD1 to 11beta-HSD2 expression, increasing conversion from active to inactive glucocorticoids. This provides a possible explanation for the antiglucocorticoid effects of DHEA.”
Note: Coffee, aspirin, and vitamin D all reduce the activity of the related 11ß-HSD1 enzyme, which works in the opposite direction, so they can help lower cortisol, too.
Ref: DHEA induces 11 -HSD2 by acting on CCAAT/enhancer-binding proteins
Jan 14

Drugs that reduce local cortisol by reducing 11β-HSD1 are being investigated for the treatment of type 2 diabetes.
Cortisol raises blood sugar and free fatty acids - essential for maintaining blood sugar in healthy states - but a driver of blood sugar dysregulation when chronically elevated.
Coffee, aspirin, and vitamin D all reduce the enzymes activity, which can help lower local cortisol regeneration. All three are associated with a reduced risk of incident diabetes (low-dose in the case of aspirin).
(11ß-HSD1 acts locally, meaning cortisol blood tests are not useful.)
“The cortisol-regenerating enzyme 11ß-HSD1 amplifies glucocorticoid levels in liver and adipose tissue. 11B-HSD1 inhibitors are being developed to treat type 2 diabetes... Conclusions: Whole-body 11B-HSD1 activity is increased in obese men with type 2 diabetes, whereas liver 11B-HSD1 activity is sustained, unlike in euglycemic obesity. This suppor the concept that inhibitors of 11ß-HSD1 are likely to be most effective in obese type 2 diabetic subjects.” PMID: 21266326

2
9
83
5,341

People sell btc to usd to pay for things too, idk why interconversion is anathema to you.
ETH is the only asset with trustless defi - ergo it is freedom money.
11
238

May 14
Then the liver receives vitamers via the portal vein and is traditionally viewed as the main conversion hub.
The main steps are:
-Uptake: Free vitamers (PL, PN, PM) enter hepatocytes.
-Salvage reactions (same enzymes as intestine):
PDXK phosphorylates: PL → PLP, PN → PNP, PM → PMP.
PNPO oxidizes: PNP → PLP, PMP → PLP.
-Regulation and storage: Liver PLP is bound to proteins (glycogen phosphorylase).
Excess PLP is dephosphorylated by phosphatases (PDXP) to PL, which can be released into circulation or further oxidized to 4-pyridoxic acid (main urinary metabolite) by aldehyde oxidase or dehydrogenase.
Export: Most circulating B6 in blood is PLP bound to albumin (via a Schiff base with lysine). To deliver to tissues, PLP is hydrolyzed back to PL by ecto-enzymes (alkaline phosphatases) on cell surfaces.
The liver also handles interconversion and catabolism, preventing toxic buildup of free PL (a reactive aldehyde).
But virtually all cells express the salvage enzymes to varying degrees, allowing local activation and recycling from degraded enzymes or circulating vitamers.
This is crucial for tissues with high PLP demand such as the brain, muscle and erythrocytes.
1
2
215

Conjugated Molecular Interconversion Accelerates Interfacial Desolvation Kinetics for Aqueous Zn Metal Batteries
advanced.onlinelibrary.wiley…
2
33

Unique Metal–Ligand Proton Tautomerism Underlying the Reversible Electrocatalytic NAD /NADH Interconversion | Journal of the American Chemical Society pubs.acs.org/doi/10.1021/jac…
3
10
1,667
