
auto-align2の対抗馬になるのかなぁ。
一旦位相合わせた後の微調整は楽そう。
デモ音源があまりによく出来すぎてるのがどっちなのか・・・。
forward-audio.com/products/p…
3
544

5/17 10AMからPaper Talkを配信します
今回は5月前半にポストした研究に関してLLMで作った概要図を使って紹介していきたいと思います
【#PaperTalk #9】 バイオインフォマティクス最新研究紹介 2026年5月前半 mTM-align2 OpenBind Genie3 ... youtube.com/live/gayaKrFlluU…
2
7
3,256
ESM-IFとcontrastive学習による埋め込みでタンパク質構造データベースを検索可能なmTM-align2が公開されました。momerは中央値30秒・multimerも90秒以内で完結し、Foldseekより高い精度と再現率を達成しています
ソースはリプ🔽
ALT 概要図は ChatGPTを用いて作成しています。 内容の理解を助けるための要約図のため、構造式、細かな数値や表現、ニュアンスに一部誤りが含まれる可能性があります。 特には構造式は大きく構造が異なる可能性が高いので、あくまでもLLMによる模式図と捉えて下さい。
1
5
16
1,186
mTM-align2: A Server for Real-time Protein Structure Database Search and Alignment
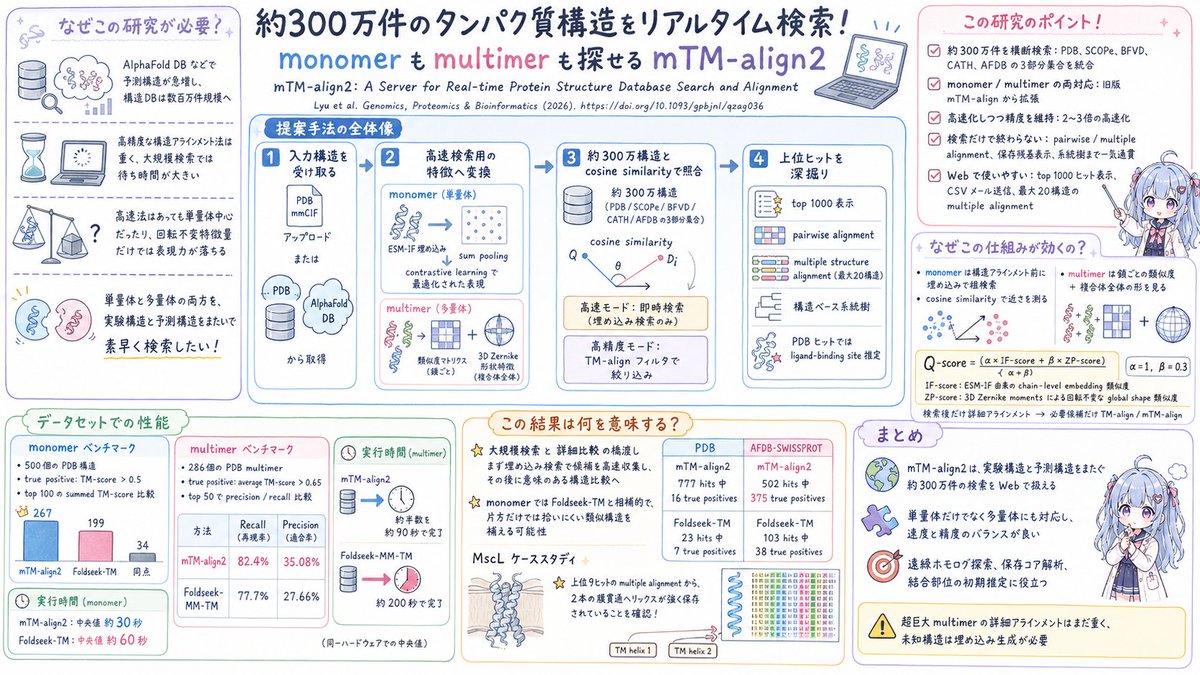
1. mTM-align2 is presented as a real-time web server that searches structurally similar proteins across ~3 million structures (experimental predicted) in seconds, while supporting both monomeric proteins and multimeric complexes.
2. The key idea is to replace expensive all-vs-all structural alignments with fast cosine-similarity search over precomputed structural fingerprints (embeddings), then optionally run detailed pairwise/multiple alignments with mTM-align for interpretability and validation.
3. For monomers, the server encodes a query using residue-level embeddings from the pretrained inverse-folding protein language model ESM-IF, aggregates them into a protein-level vector (sum pooling), and uses contrastive learning so cosine similarity correlates with structural similarity.
4. For multimers, it combines (a) chain-wise monomer embeddings and (b) a rotation-invariant global shape descriptor based on 3D Zernike moments; the final similarity (Q-score) is a weighted sum of chain similarity (IF-score) and shape similarity (ZP-score), with weights α=1 and β=0.3.
5. mTM-align2 integrates multiple major databases in one interface: PDB (including large monomer and multimer sets), SCOPe and CATH domain databases, BFVD viral structures, plus several AlphaFold DB subsets (Swiss-Prot, organism set, global health set), enabling cross-database retrieval rather than siloed searches.
6. The server offers two search modes: a high-accuracy mode that adds a fast TM-align-based filtering step to improve precision, and a high-speed mode that skips filtering for near-instant results; results are returned as ranked hits (top 1000) and can be emailed as CSV for batch workflows.
7. Beyond retrieval, the output workflow is designed for downstream analysis: users can launch pairwise superposition with TM-align metrics (TM-score, RMSD) and residue-level correspondence, or run multiple structure alignment (up to 10 selected hits via the UI) to identify conserved cores and generate a structure-based phylogenetic tree.
8. A practical annotation feature is included for PDB hits: after superposition, the server transfers ligand context from Q-BioLiP and marks query residues within 5 Å of the aligned ligand as putative binding residues, providing fast template-based binding-site hints (with the paper noting it is not meant to replace specialized binding-site predictors).
9. Benchmarks against Foldseek variants suggest complementary strengths for monomer search (mTM-align2 wins on more test cases by summed true-positive TM-scores in top-100, while Foldseek leads on others), and stronger multimer retrieval performance versus foldseek-multimer (top-50 recall ~82.4% vs 77.7%; precision ~35.08% vs 27.66%).
10. A case study on the MscL mechanosensitive channel highlights sensitivity to remote homologs and conformational diversity: against AFDB-SwissProt, mTM-align2 retrieves many more true positives than Foldseek, and multiple alignment of top hits reveals strictly conserved transmembrane helices linked to gating architecture.
💻Code: ngdc.cncb.ac.cn/biocode/tool…
📜Paper: doi.org/10.1093/gpbjnl/qzag0…
#ProteinStructure #StructuralBioinformatics #AlphaFold #ProteinComplexes #StructureSearch #BioinformaticsTools #ESM #ContrastiveLearning #ZernikeMoments #TMAlign

5
39
2,642

録りの時はAvidのTime AdjusterをDSPで使って合わせて、
録り終わったらauto-align2で位相を確かめて、その結果をTime Adjusterに反映させます。
2...
続き→marshmallow-qa.com/messages/…
#マシュマロを投げ合おう
11
1,278

Feb 25
Alignmentもう一度試してDrum GATE2 最高!と思ったのだけど、 もう少し試したらどうしてもAuto-Align2の方が結果が良かった😅。 ので現時点ではAuto-Align2から普通にDrum gate とか使ってます。(時間は多少かかりますが)
まあ一番大事なのはドラマーの腕です😉
2
189

Jan 27
pov me yg ngerjain powerpoint nambah shortcut align2 sama textbox :"
Hidup ALT
Jan 26

atasanku, abis ngeliat aku ngerjain laporan excel tanpa mouse. terus dia amaze, semoga beban kerjaku abis itu gak ditambah yaa 🙃🙏
4
664

Jan 19
Trending every episode,tapos align2 lang fan service sa amin 🤭😅
Pero ayos lang,ginusto nmn to eh 🫠

More than deserving like hello, hindi pa tapos ang series 10k plus yung rating sa @VivaOnePH app. Baka naman po oh. Masakit na kamay namin kakalampag eh 🤣😅✌️
2
4
23
321

31 Oct 2025
うーんベースを2本レイヤーしたいのだが、位相がズレて音がよれてしまう。
手作業で合わせるかね。Auto Align2セールきたら買おうかな
1
2
332


22 May 2025
Rapid and accurate protein structure database search using inverse folding model and contrastive learning
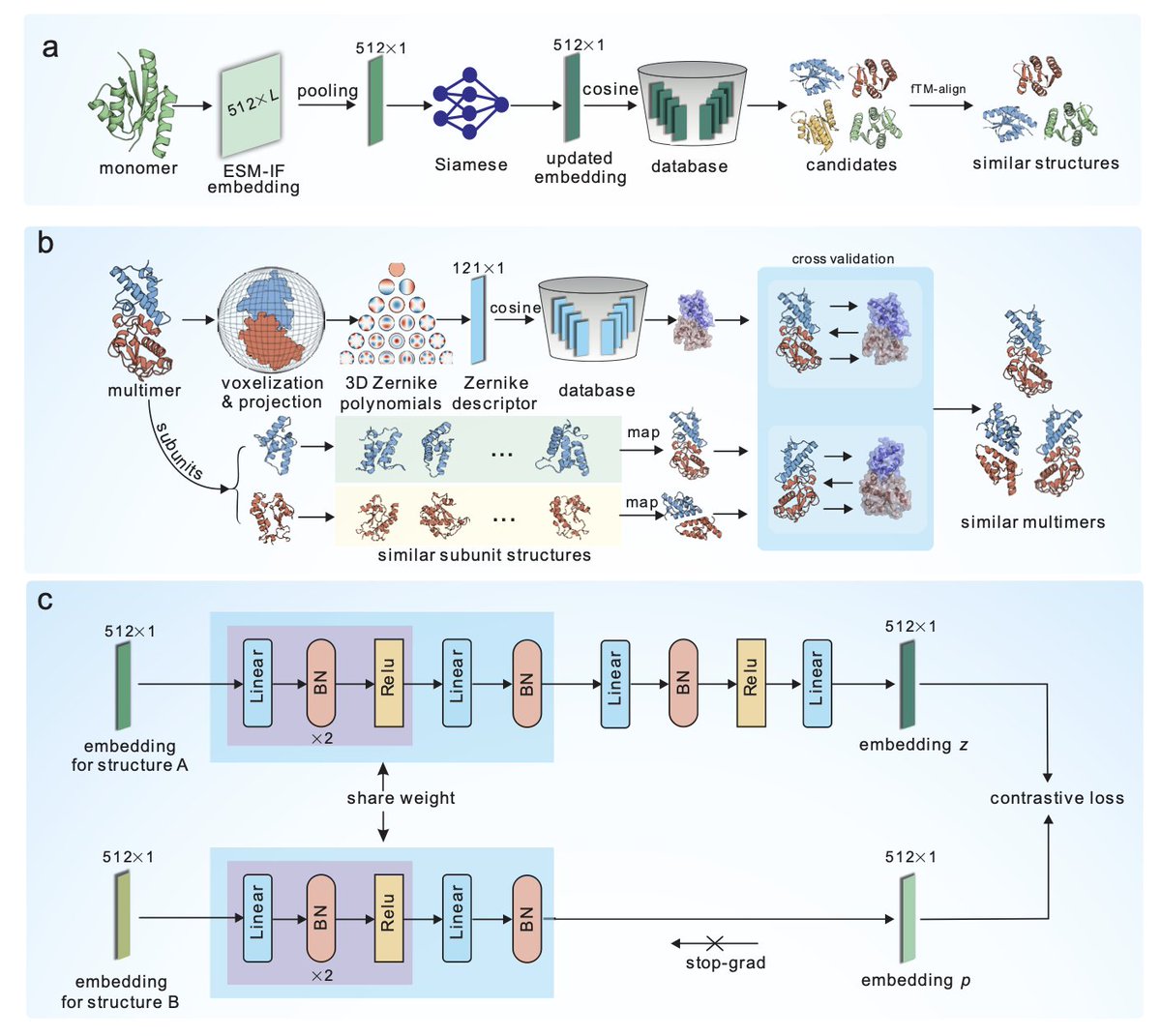
1.mTM-align2 is a fast and accurate protein structure search framework that uses embeddings from an inverse folding model (ESM-IF) optimized by contrastive learning. It enables structure retrieval across large protein databases with high precision and speed, outperforming existing methods on both monomers and multimers.
2.The method converts 3D protein structures into 512-dimensional embeddings via ESM-IF, followed by refinement with a Siamese contrastive learning network trained on over 7 million structure pairs. This embedding captures structural similarity directly, enabling cosine-based similarity scoring.
3.For monomeric structures, mTM-align2 completes searches within seconds and achieves over 90% precision for top 10 hits. It is significantly faster than Foldseek, yet competitive or superior in recall and sum TM-score metrics across various test sets.
4.Multimeric structure search is supported via a dual-module system: one uses 3D Zernike polynomials to capture global shape, while the other decomposes multimers into monomeric subunits processed through the ESM-IF pipeline. Results from both modules are fused via a weighted Q-score.
5.In benchmarks against 286 multimeric queries, mTM-align2 outperforms Foldseek-MM-TM in both precision and recall. At a TM-score threshold of 0.5, mTM-align2 achieves up to 97.14% top-10 precision, demonstrating its robustness even for novel or complex structures.
6.SCOPe classification tests show that mTM-align2 embeddings align well with domain-level protein structure definitions, maintaining high precision across fold, superfamily, and family levels. Removing structure-based filters reduces performance, confirming the necessity of both embedding and alignment stages.
7.t-SNE visualizations of over 15,000 SCOPe structures show that mTM-align2 embeddings effectively cluster proteins by structural class. The model also reveals likely misclassifications and ambiguous definitions in SCOPe, suggesting its potential for aiding structural annotation.
8.On AlphaFold DB, mTM-align2 accurately clusters high-confidence structures across five diverse InterPro domains. It can detect sub-clusters corresponding to multi-domain or oversized proteins, highlighting its capacity to handle real-world structural variability.
9.Ablation studies confirm that both the Siamese contrastive learning module and structure alignment filtering are critical to performance. Removing either component significantly degrades precision, demonstrating their synergistic effect.
10.mTM-align2 integrates deep learning with classic structure alignment in a scalable pipeline. Its speed, precision, and broad applicability make it a powerful tool for structural bioinformatics, with a web server publicly available for use.
📜Paper: biorxiv.org/content/10.1101/…
#ProteinStructure #StructuralBioinformatics #InverseFolding #ContrastiveLearning #TMAlign #Foldseek #AlphaFold #ComputationalBiology #mTMAlign2

2
7
53
4,054
22 May 2025
Rapid and accurate protein structure database search using inverse folding model and contrastive learning
1.mTM-align2 is a fast and accurate protein structure search framework that uses embeddings from an inverse folding model (ESM-IF) optimized by contrastive learning. It enables structure retrieval across large protein databases with high precision and speed, outperforming existing methods on both monomers and multimers.
2.The method converts 3D protein structures into 512-dimensional embeddings via ESM-IF, followed by refinement with a Siamese contrastive learning network trained on over 7 million structure pairs. This embedding captures structural similarity directly, enabling cosine-based similarity scoring.
3.For monomeric structures, mTM-align2 completes searches within seconds and achieves over 90% precision for top 10 hits. It is significantly faster than Foldseek, yet competitive or superior in recall and sum TM-score metrics across various test sets.
4.Multimeric structure search is supported via a dual-module system: one uses 3D Zernike polynomials to capture global shape, while the other decomposes multimers into monomeric subunits processed through the ESM-IF pipeline. Results from both modules are fused via a weighted Q-score.
5.In benchmarks against 286 multimeric queries, mTM-align2 outperforms Foldseek-MM-TM in both precision and recall. At a TM-score threshold of 0.5, mTM-align2 achieves up to 97.14% top-10 precision, demonstrating its robustness even for novel or complex structures.
6.SCOPe classification tests show that mTM-align2 embeddings align well with domain-level protein structure definitions, maintaining high precision across fold, superfamily, and family levels. Removing structure-based filters reduces performance, confirming the necessity of both embedding and alignment stages.
7.t-SNE visualizations of over 15,000 SCOPe structures show that mTM-align2 embeddings effectively cluster proteins by structural class. The model also reveals likely misclassifications and ambiguous definitions in SCOPe, suggesting its potential for aiding structural annotation.
8.On AlphaFold DB, mTM-align2 accurately clusters high-confidence structures across five diverse InterPro domains. It can detect sub-clusters corresponding to multi-domain or oversized proteins, highlighting its capacity to handle real-world structural variability.
9.Ablation studies confirm that both the Siamese contrastive learning module and structure alignment filtering are critical to performance. Removing either component significantly degrades precision, demonstrating their synergistic effect.
10.mTM-align2 integrates deep learning with classic structure alignment in a scalable pipeline. Its speed, precision, and broad applicability make it a powerful tool for structural bioinformatics, with a web server publicly available for use.
📜Paper: biorxiv.org/content/10.1101/…
#ProteinStructure #StructuralBioinformatics #InverseFolding #ContrastiveLearning #TMAlign #Foldseek #AlphaFold #ComputationalBiology #mTMAlign2
1
2
552

20 Mar 2025
先日Avid Pro Tools StudioがDolby内部レンダラーを標準搭載していたことに驚き慌ててアップデートしたが、ARAというMelodyneなどオーディオの読み込み不要で使えるようになる機能、なんだか目から鱗と思われる機能が最近満載。
SoundRedax社のAuto-Align2(位相修正プラグイン)も気になるー!
1
2
356

16 Jan 2025
これってauto align2のギター専用みたいなやつなんですかね…??
16 Jan 2025

ギタリストでMixする人、ギターロックめちゃくちゃMixする人、全員持ってて損しないプラグインです。ようやくApple Silicon対応してくれて、もう本当にこれで100%、Rosettaを使用してなんちゃら、みたいなのは使わないし未練も無いです♩
2
600



1 Dec 2024
今度のドラムレコーディングで大活躍する(予定) 自動位相合わせプラグイン Auto-Align2がセール中!
Auto-Align: The Automatic Phase Alignment Plug-in | Sound Radix soundradix.com/products/auto…
1
4
483

29 Oct 2024
Auto Align2が気になってたんだけど、これ手持ちのneutron4から5にアップデートしたらphase moduleでおんなじことできそうだなあ
izotopeさんwaves update planみたいに手持ちのプラグイン一括でアップデートできるライセンスとか作ってほしい……
1
4
486

このタイミングでPro ToolsにAuto-Align2がARA統合された事に気づいて超ハッピー。プラグインだとレイテンシーがすごいけど、ARAなら関係ないからレコーディング中にもバリバリ使っている!
10
974

21 Jun 2024
auto-align2のARA対応
元々インサートだとそこそこプラグインディレイあるけど、ARA使うとそこにはディレイ発生しないメリットが。
ただARAだと一部をキャプチャーして解析って訳にはいかなくてトラック丸っとになるからライブ物だと解析時間がまあまあかかるデメリット🧐
#autoalign2 #protools #ARA
6
662