
Jun 8
🚀 REGISTRATION IS OFFICIALLY OPEN FOR EPISODE 1!
We are kicking off our flagship ISCB-SC & EMBL-EBI EduSearch series with PDBe-KB, a global webinar series with the ultimate paradigm-shifter in computational proteomics: The AlphaFold Database. Before DeepMind’s AI breakthrough, determining a single protein’s 3D structure could consume an entire wet-lab PhD pipeline. Today, AlphaFold provides access to over 200 million structural predictions. But are you utilizing its full analytical potential for your research projects?
If you are a student, researcher, or biotechnology enthusiast looking to leverage AI-powered structural models for mutation analysis, drug discovery, or wet-lab validation, this masterclass is built for you.
🎯 WHAT YOU WILL MASTER IN THIS SESSION: 1.Navigating the AlphaFold DB interface like an expert. 2.Interpreting prediction confidence metrics critically (pLDDT scores & PAE alignment matrices). 3.Seamlessly extracting structural metadata to power your research manuscripts.
📅 Save the Date: 26th June,2026
⏰ Global Time Zones (Synchronized):
• 🇮🇳 IST (India): PM🇵🇰 PKT (Pakistan): PM
• 🇧🇩 BST (Bangladesh): PM
• 🇹🇷 TRT (Turkey): PM
• 🇳🇬 WAT (Nigeria): AM
• 🇰🇷 KST (South Korea): PM
🎟️ Seats are limited across our 6 participating chapters. Secure your international certificate eligibility early! 🔗 REGISTER NOW VIA THE LINK BELOW: forms.gle/3JNnHHS6WCyLXF5F8 #Bioinformatics #AlphaFold #DeepMind #ProteinFolding #StructuralBioinformatics #Proteomics #DrugDiscovery #Genomics #WebinarAlert #RSGs #Biotechnology #AIInHealthcare
@iscb @RSG_nigeria @rsg_pak @ISCB_RSG_India @RSGBangladesh @RSG_KOREA @emblebi

3
7
311

AF Cache: Efficient Pipeline for Running AlphaFold for High-Throughput Protein-Protein Interaction Prediction
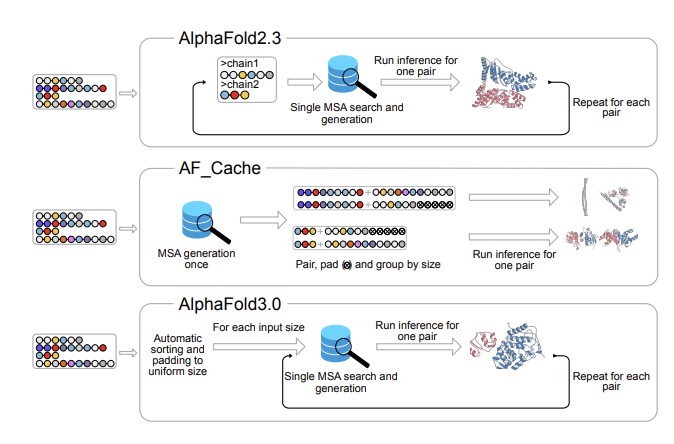
1 AF Cache is a Nextflow pipeline designed to make large-scale protein–protein interaction (PPI) screening practical with AlphaFold2 and AlphaFold3 by cutting the dominant runtime bottlenecks: repeated MSA generation and (for AF2) repeated JAX recompilations.
2 The key idea is to generate MSAs once per unique monomer for the whole dataset, then reuse (“cache”) them across all pairs. For an all-against-all screen of N proteins, default multimer workflows can trigger ~N(N 1) chain-level MSA generations, while AF Cache reduces this to N by construction.
3 AF Cache accelerates MSA generation by using GPU-accelerated MMseqs2 and batching all proteins in a single dataset-level step (rather than per-target or small batches). It also overlaps CPU and GPU steps across UniRef and environmental database alignments to reduce GPU idle time.
4 For AlphaFold2 specifically, AF Cache adds sequence-length bucketing for multimers (bucketed by total chain length) and pads within each bucket so JAX compilation happens once per bucket instead of once per pair—saving ~1–2 minutes of GPU time for every additional multimer in that bucket.
5 Benchmark: 100 human mitochondrial proteins (40–1000 aa) were screened all-against-all including homodimers (5,050 pairs). Settings were streamlined for throughput (AF2: single model, 3 recycles, templates disabled; AF3: single seed, one diffusion sample).
6 Pre-prediction speedups (MSA stage): when comparing to a realistic 128-CPU-core baseline, AF Cache achieved ~13x faster MSA generation for AF2 settings using full BFD, and ~5x faster for AF3 settings using small BFD. The paper also reports very large raw GPU-vs-CPU core-hour reductions, emphasizing the benefit of GPU-based MSA search in high-throughput regimes.
7 End-to-end impact for AF2: caching bucketing reduced AF2 prediction compilation time from 253 to 125 GPU hours (>50% reduction), and made inference ~90 seconds faster per protein pair on average while preserving the same runtime scaling with pair length.
8 Output similarity was assessed via ipTM comparisons. Correlations between default vs AF Cache runs were moderate across all pairs (Pearson r ~0.70 for AF2; ~0.64 for AF3), but much higher for pairs where both proteins map to a shared PDB entry (r ~0.98 for AF2; ~0.94 for AF3), suggesting structurally supported cases are highly consistent across pipelines.
9 Practical deployment details: AF Cache provides a ready-to-use workflow for local and HPC environments, supports all-against-all or user-specified pair lists, automates AF3 JSON preparation, and can download/install dependencies (including databases; AF2 network parameters) as needed.
💻Code: github.com/clami66/AF_cache
📜Paper: arxiv.org/abs/2606.04566
#AlphaFold #ProteinInteractions #PPI #Bioinformatics #ComputationalBiology #StructuralBioinformatics #Nextflow #HPC #MMseqs2 #Proteomics
14
75
21,239
DeepRank-Ab: A scoring function for antibody-antigen complexes based on geometric deep learning
1. DeepRank-Ab targets a key failure mode in antibody–antigen modeling: good poses are often sampled but not ranked correctly. The paper shows an example where AlphaFold3 generates a DockQ 0.6 model yet ranks it 498th, illustrating that scoring (not only sampling) is a central bottleneck.
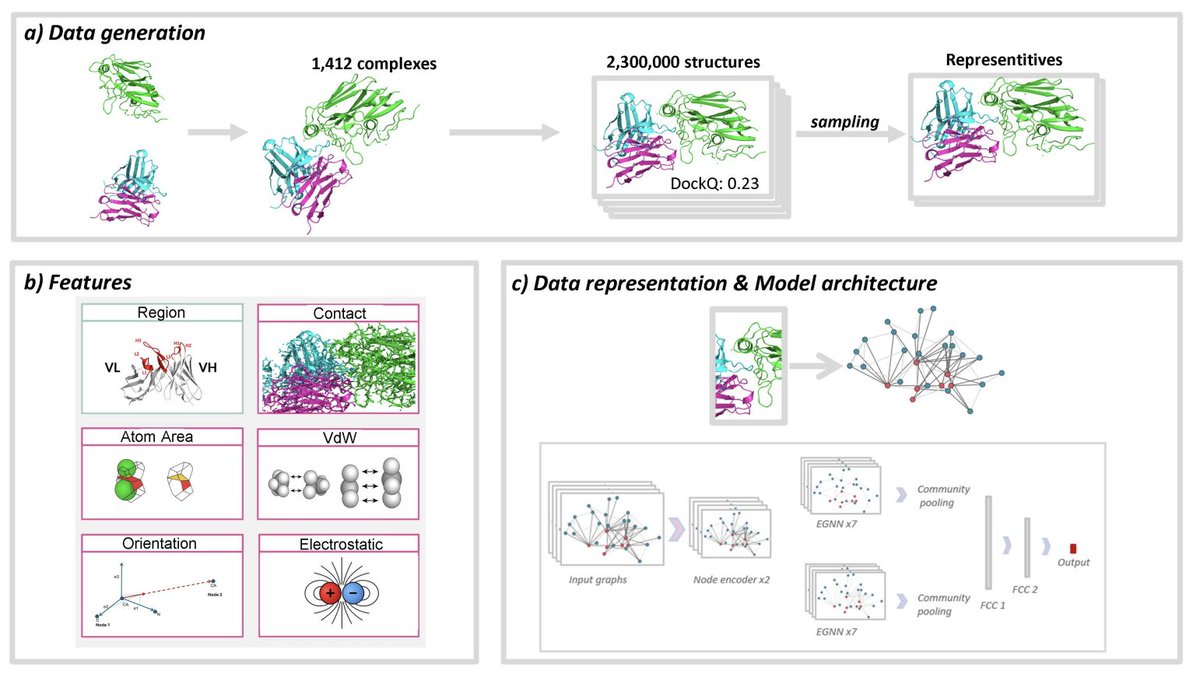
2. The authors introduce a rigorously curated benchmark tailored to antibody–antigen interfaces: ~2.3 million decoys from 1,442 complexes (from SAbDab), generated under four HADDOCK3 protocols (bound/unbound docking and refinement). Complexes are clustered with Foldseek-Multimer and split at the cluster level to reduce data leakage.
3. DeepRank-Ab is trained as a regression model to predict continuous DockQ (pDockQ), rather than a binary “acceptable/not acceptable” label. This preserves the full spectrum of model quality and directly supports ranking.
4. A major design finding: atom-level interface graphs outperform residue-level graphs for antibody–antigen scoring, consistent with the importance of fine-grained CDR loop geometry. The best-performing formulations combine atom-level nodes with interface-focused edges.
5. The interface representation is strengthened using Voronoi-based surface decomposition: edges can encode either simple interatomic distances or Voronoi contact areas. Across cross-validation, Voronoi contact area provides a consistent edge over distance-only encoding for predicting and classifying near-native poses.
6. Antibody-specific and physics-inspired features matter. Adding IMGT-derived region labels (e.g., CDR annotations) and geometric/energetic descriptors improves performance and stability; ablations show large error increases when removing region labels, atom type, covalent interactions, orientation, or Voronoi contact area. In contrast, raw ESM-2 embeddings are partly redundant, and antibody-specific fine-tuning (AbTune) yields only marginal gains relative to added complexity.
7. Training data sampling is treated as a first-class modeling choice. Besides “balanced” sampling across DockQ bins, they test targeted upsampling of low-quality decoys (DockQ < 0.23) to increase structural diversity in failure modes; this slightly improves overall learning and robustness.
8. Architecture: an E(n)-equivariant GNN with a two-branch design that processes interface edges and internal edges separately, then pools with global attention and predicts pDockQ. This keeps rotational/translational symmetry while learning local interface geometry.
9. On the hardest docking benchmark setting (unbound–unbound docking; n=215), DeepRank-Ab variants outperform HADDOCK EMscoring and VoroIF-jury in Top-k success. Atom-distance edges do best at Top1, while Voronoi-area edges become strongest at higher k (e.g., Top10), reflecting tradeoffs between sharp early ranking and broader enrichment.
10. Generalization beyond docking decoys: on an AF3-focused external test set (59 complexes released after AF3’s training cutoff), DeepRank-Ab improves AF3 Top1 success by 35.5% and more than doubles mean Top1 DockQ. A notable practical tweak is removing the electrostatics term to better handle unrelaxed AF3 structures (distribution shift from clashes/packing), boosting Top1 success to 54.24%.
11. External CAPRI/MassiveFold evaluation (5 antibody–antigen targets) suggests strong out-of-distribution performance: DeepRank-Ab reaches 100% Top5 success, with especially strong results on peptide antigens. The paper also analyzes why: peptide interfaces tend to be more locally driven and compatible with the model’s 5 Å heavy-atom cutoff graph construction.
12. The work argues that “consensus scoring” is not automatically beneficial: combining DeepRank-Ab with AF3 via averaging or jury voting reduces performance, implying DeepRank-Ab already captures much of the useful ranking signal for these systems.
💻Code: github.com/haddocking/DeepRa…
📜Paper: doi.org/10.1038/s42003-026-1…
#ComputationalBiology #StructuralBioinformatics #Antibodies #ProteinDocking #GeometricDeepLearning #GNN #AlphaFold3 #HADDOCK #CAPRI #BenchmarkDatasets
8
49
3,005
Atom-level Protein Representation Learning Improves Protein Structure Prediction
1. The paper proposes TRIPROREP, a protein representation model that explicitly targets structure-predictive use cases (complex docking signals and structure-model supervision), rather than only downstream function annotation where sequence-only PLMs can remain competitive.
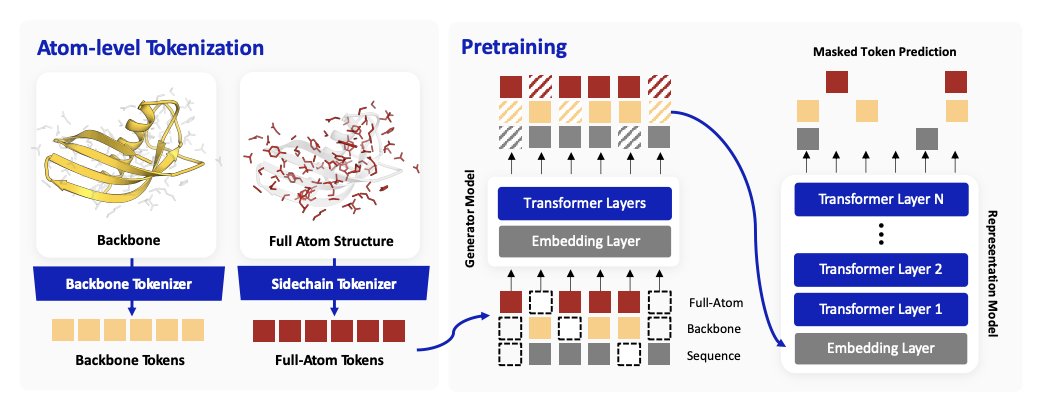
2. Core idea: learn a coupled residue-level representation from three aligned “views” of the same protein: amino-acid identity, backbone geometry tokens, and newly introduced full-atom residue geometry tokens that preserve side-chain/heavy-atom arrangements important for interfaces.
3. The full-atom tokenization is built with a residue-level VQ-VAE over Atom37 heavy-atom coordinates expressed in an SE(3)-invariant local frame (defined by N, Cα, C), plus dihedral/torsion features; a 512-entry codebook captures rotamer-sensitive local atomic states that backbone-only tokens largely miss.
4. Pretraining objective: ELECTRA-style generator corruption across all three token streams, followed by a discriminator trained to recover the original tokens (not just “replaced vs unchanged”). Independent corruption per view creates plausible-but-inconsistent cross-view combinations, forcing the model to learn cross-view consistency between sequence, backbone, and side-chain geometry.
5. Scale and data: TRIPROREP is pretrained at 35M, 150M, 650M, and 2.8B parameters on 83.7M predicted structures from AFDB (filtered by pLDDT) plus ESMAtlas, using 512-residue crops and a high masking probability per view to amplify corrective signals.
6. The paper also introduces REPSP, a structure-predictive benchmark built from 1.8M AFDB homodimer complexes (paired with corresponding apo monomers), split by 50% sequence-identity clusters, designed to test whether representations contain geometry useful for predicting complexes and supervising folding models.
7. REPSP Task 1 (homodimer co-folding / flexible docking): a modified SimpleFold-style flow-matching docking model takes frozen apo-chain representations and predicts holo homodimer structures. TRIPROREP improves interface and overall metrics across all sizes (e.g., 2.8B reaches DockQ 0.499 vs ESM2-3B 0.387; 650M already beats several “huge” baselines on most metrics).
8. REPSP Task 2 (per-residue interface property probing): simple MLP probes on frozen monomer representations predict homodimer-derived labels (binding site, ∆SASA, Levy tier, bond/interaction types). TRIPROREP consistently improves AUPRC/AUROC and correlation metrics, suggesting binding-relevant signals are encoded at residue resolution without fine-tuning.
9. REPSP Task 3 (representation-aligned distillation for monomer folding): pretrained representations are used as dense alignment targets (cosine similarity) to guide a structure predictor’s hidden states. TRIPROREP provides the strongest alignment target among large-scale models at 650M (e.g., TM-score 0.5822 vs baseline 0.5422), indicating its geometry is not only decodable but also useful as supervision.
10. Generalization and trade-offs: gains persist on real-world RCSB homodimers deposited after June 2023, and TRIPROREP remains competitive (not necessarily best) on conventional EC/GO function probing—supporting the paper’s thesis that structure-aware pretraining should be evaluated on structure-predictive tasks, not only function benchmarks.
💻Code: holymollyhao.github.io/TriPr…
📜Paper: arxiv.org/abs/2605.22133
#ProteinRepresentationLearning #ProteinStructure #StructuralBioinformatics #ComputationalBiology #DeepLearning #GenerativeModels #ProteinDocking #AlphaFold #ESMFold #VQVAE
9
53
2,727
PockFlex: A web server for flexibility-aware binding site identification and prioritisation from structural ensembles
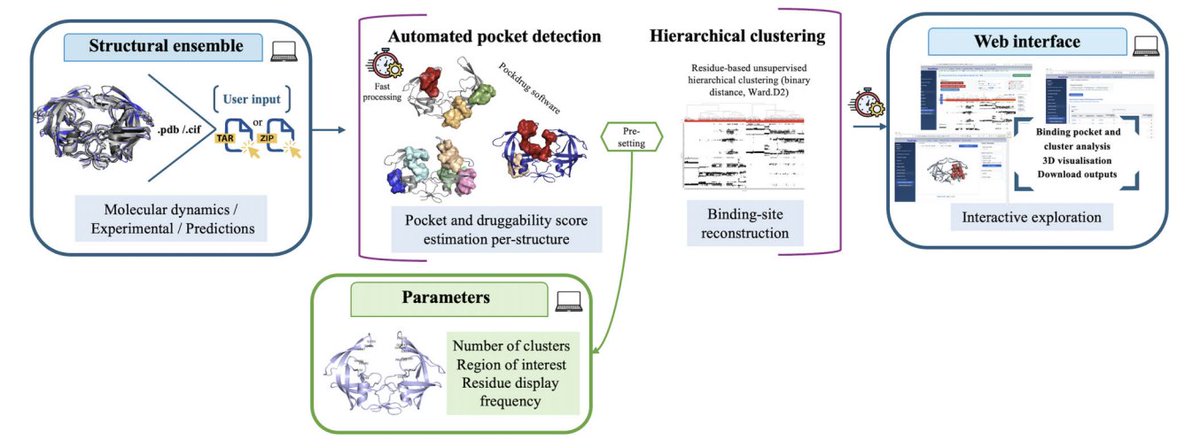
1. PockFlex is a free, no-login web server that finds and prioritises binding sites across structural ensembles (MD trajectories, multiple experimental structures, or predicted conformations), explicitly treating conformational variability as a first-class signal rather than noise.
2. Core idea: detect pockets independently in each conformation, then reconstruct “recurrent binding site organisations” by clustering pockets across the ensemble using residue-level similarity (which residues line the pocket), enabling persistent vs transient/rare pocket states to be separated.
3. A key practical design choice is the user-defined region of interest (ROI): not a predefined binding site, but an exploration scope (a residue set). PockFlex keeps only pockets overlapping the ROI, supporting iterative workflows: start broad, then refine ROI and clustering resolution once recurrent clusters appear.
4. Methodologically, the ensemble-level grouping uses binary residue fingerprints and an asymmetric binary distance (appropriate for presence/absence), followed by hierarchical clustering (Ward.D2). This residue-centred approach is meant to better track conserved pockets/subpockets under flexibility than comparing global pocket descriptors alone.
5. The server integrates pocket-level druggability from the established PockDrug workflow (Fpocket-based pocket estimation druggability scoring). Importantly, PockFlex does not replace pocket-level scores: it summarises them at the cluster level as the fraction of conformations in that cluster predicted druggable (score_d ≥ 0.50), while keeping per-pocket heterogeneity accessible.
6. Output focuses on interpretability for ensemble analysis: dendrograms residue–pocket heatmaps to inspect cluster coherence, residue-frequency profiles to quantify which residues are stable vs variable within a cluster, and interactive 3D visualisation to map clusters onto structures.
7. Multi-resolution exploration is built in via the parameter K (number of clusters). PockFlex proposes a heuristic starting K (average number of ROI-overlapping pockets per conformation), but encourages users to tune K to either split flexible sites into sub-states or merge closely related pocket organisations.
8. Example (HIV-1 protease, 500 ns MD; 501 frames): thousands of pockets were detected and clustered, recovering the central active-site region without predefining it, while also separating multiple central-site conformational states consistent with flap dynamics and highlighting recurrent lateral pockets (e.g., outer-surface regions near fulcrum/wall) with non-trivial recurrence across frames.
9. Implementation notes relevant for reproducibility/usage: Django PostgreSQL backend, R-based workflow for clustering, containerised with Docker on the server side; accepts up to 500 conformations, ROI up to 300 residues, 100 MB compressed upload; typical runtime reported as ~1.2 s per structure for pocket detection plus ~1–2 min clustering for standard jobs.
📜Paper: doi.org/10.1093/nar/gkag453
#computationalbiology #structuralbioinformatics #drugdiscovery #proteindynamics #moleculardynamics #bindingsites #druggability #webserver #bioinformatics #NAR
8
32
2,710
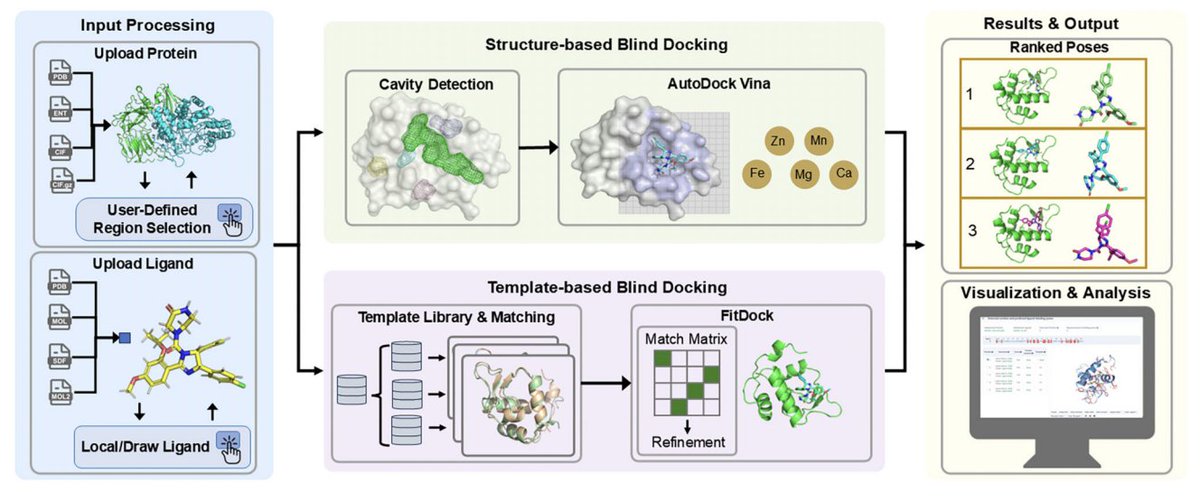
CB-Dock3: An Enhanced Web Server for Protein–Ligand Blind Docking
1. CB-Dock3 is a major update of the CB-Dock blind docking web server, targeting the practical bottlenecks created by today’s surge of large Cryo-EM and AI-predicted protein structures (e.g., AlphaFold3-era multi-domain complexes).
2. On the CASF-2016 benchmark (285 complexes), CB-Dock3 reaches a 67.4% success rate (top-ranked pose RMSD ≤ 2.0 Å), a 10.6 percentage-point absolute improvement over CB-Dock2 (56.8%), and higher than several commonly used blind docking alternatives evaluated in the paper.
3. The core algorithmic upgrade is an improved template-guided docking engine (FitDock1.2) integrated with BioLiP2 (2025.04.23), whose biologically relevant protein–small molecule and protein–peptide interactions are reported to be nearly doubled vs the prior template resource.
4. FitDock1.2 uses hierarchical six-layer atomic feature matching to enforce chemically consistent template-to-ligand alignment, designed to avoid connectivity conflicts seen in earlier multi-point matching and to maintain structural continuity during guided docking.
5. A key practical chemistry improvement is stereochemical feature transfer: beyond torsion sampling, the engine explicitly adjusts bond angles and enforces chiral centers in the matched substructure to remain consistent with the template, helping prevent local geometry drift (notably for elongated/flexible ligands such as peptides).
6. CB-Dock3 adds metal-aware and heteroatom-inclusive docking: instead of discarding HETATM records by default, it lets users selectively retain functional metal ions/cofactors, and incorporates metal-aware modeling in both AutoDock Vina 1.2.0 and FitDock (with expanded atom-type recognition and metal interaction terms trained using metal-containing complexes).
7. The server introduces a “Divide-and-Conquer” partitioning workflow for massive targets: a graph-based clustering method partitions large proteins into compact regions to reduce search space and false positives; users can also define docking regions by chains/residue ranges or specify key residues to focus the search.
8. Modern structure handling is improved with native PDBx/mmCIF support (and .gz inputs), addressing PDB format size limitations; the web UI also provides interactive setup (region selection, optional metal/hetero retention, ligand upload/drawing) and result inspection (scores, contacts, 3D visualization).
9. A metalloprotein case study (human secreted phospholipase A2 group IIE, PDB 5WZU) illustrates the metal-aware benefit: keeping the catalytic Ca2 yields a much closer pose (RMSD 0.244 Å) than excluding it (RMSD 1.102 Å), consistent with preserving native coordination environments.
10. The authors emphasize intended use: CB-Dock3 is positioned for exploratory binding-site/pose hypothesis generation when sites are unknown and for deriving pocket parameters for downstream targeted docking; limitations include dependence on available homologous templates and a rigid-receptor approximation.
📜Paper: doi.org/10.1093/nar/gkag417
#BlindDocking #MolecularDocking #ProteinLigand #CADD #DrugDiscovery #StructuralBioinformatics #Metalloproteins #AlphaFold3 #CryoEM #WebServer
3
18
1,591
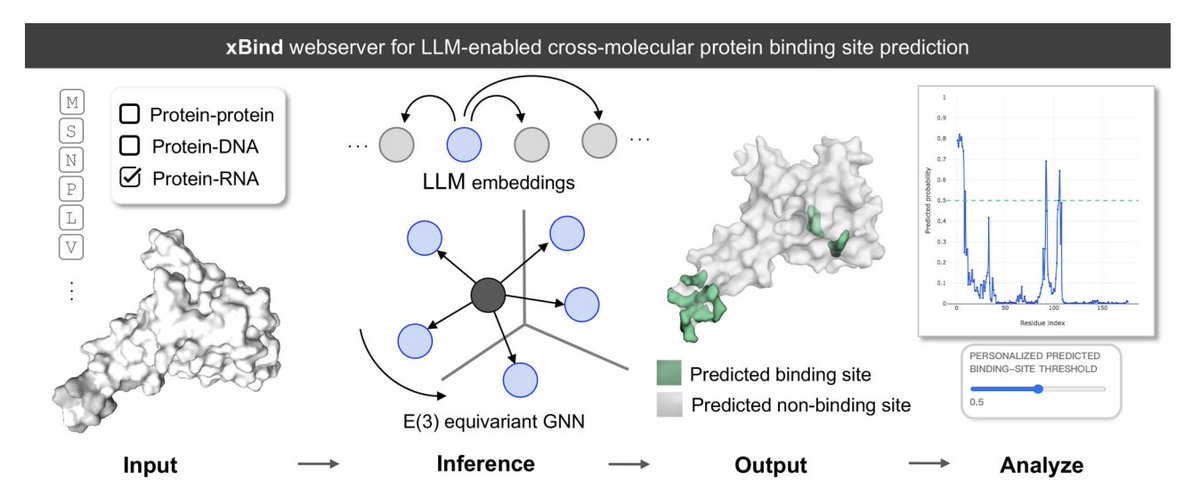
xBind: An Integrated Webserver for Large Language Model-Enabled Cross-Molecular Protein Binding Site Prediction
1. xBind is a unified webserver for residue-level binding-site prediction across three interaction types: protein–protein, protein–DNA, and protein–RNA, aiming to remove the usual “one tool per modality” fragmentation.
2. The core modeling idea is to combine ESM-2 protein language model embeddings with sequence- and structure-derived features, then learn on 3D graphs using symmetry-aware E(3)-equivariant GNNs to better respect molecular geometry.
3. A practical highlight is that xBind accepts either (a) a single-chain protein sequence (FASTA) or (b) a monomer structure (PDB/mmCIF). For sequence-only inputs, it can run AlphaFold2 on the fly to produce a structure for structure-aware inference.
4. Under the hood, xBind exposes two independently trained frameworks: EquiPPIS for protein–protein interfaces and EquiPNAS for protein–DNA/RNA interfaces, both designed to be robust to imperfect structures (including AlphaFold models).
5. Benchmarking emphasizes performance on AlphaFold2-predicted structures, reflecting real-world usage where experimental complexes are missing. Reported comparisons show statistically significant gains (95% confidence, P < 0.05) over strong baselines using ROC-AUC and PR-AUC across multiple test sets.
6. Example performance (AlphaFold inputs): for protein–protein, EquiPPIS improves over GraphPPIS on Test_60 (ROC-AUC 0.7912 vs 0.7667; PR-AUC 0.4516 vs 0.3956). For protein–DNA, EquiPNAS leads on Test_181 (ROC-AUC 0.9159; PR-AUC 0.3717) and Test_129 (ROC-AUC 0.9387; PR-AUC 0.569). For protein–RNA, EquiPNAS improves over GraphBind on Test_117 (ROC-AUC 0.8856 vs 0.7942; PR-AUC 0.3118 vs 0.2019).
7. The web UI is built for interpretability and calibration: it provides interactive sequence views, 3D visualization of predicted interface residues, per-residue likelihood plots, and user-adjustable probability thresholds to tune precision/recall for different downstream needs.
8. Output artifacts are designed for reuse: downloadable structure used for prediction, residue-level probability files, and a packaged archive with complete outputs and run metadata; jobs can be public or private, with optional email notification.
9. The paper also outlines near-term directions: calibrating predictions using residue-wise uncertainty signals (e.g., experimental B-factors or AlphaFold pLDDT) and incorporating conformational dynamics/multistate behavior to better model binding under structural heterogeneity.
📜Paper: doi.org/10.1093/nar/gkag425
#Bioinformatics #ComputationalBiology #ProteinScience #StructuralBioinformatics #DeepLearning #GraphNeuralNetworks #ProteinLanguageModels #ESM2 #AlphaFold #ProteinInteractions #NARWebServerIssue
3
1,336
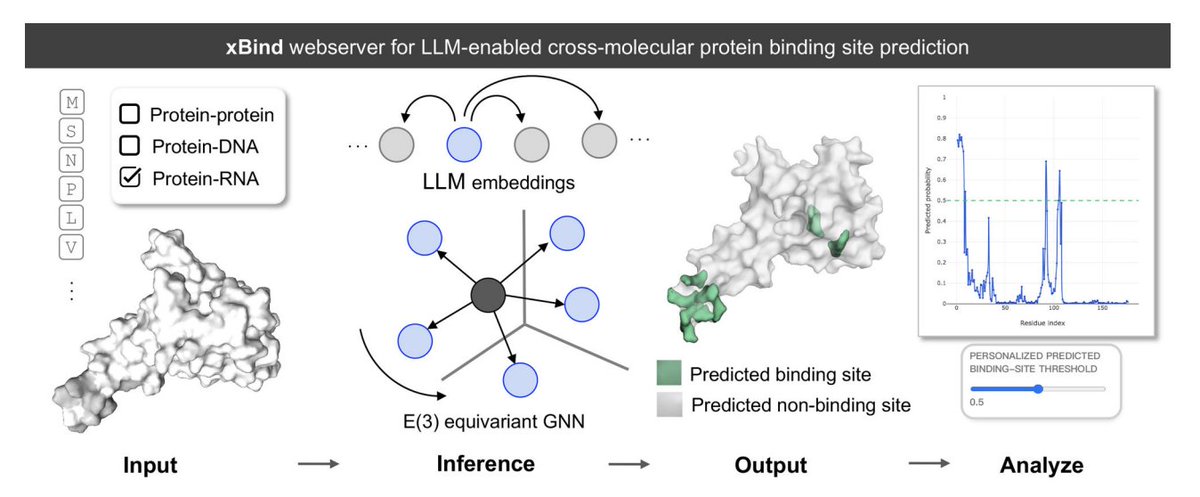
xBind: An Integrated Webserver for Large Language Model-Enabled Cross-Molecular Protein Binding Site Prediction
1. xBind is a unified webserver for residue-level binding-site prediction across three interaction types: protein–protein, protein–DNA, and protein–RNA, aiming to remove the usual “one tool per modality” fragmentation.
2. The core modeling idea is to combine ESM-2 protein language model embeddings with sequence- and structure-derived features, then learn on 3D graphs using symmetry-aware E(3)-equivariant GNNs to better respect molecular geometry.
3. A practical highlight is that xBind accepts either (a) a single-chain protein sequence (FASTA) or (b) a monomer structure (PDB/mmCIF). For sequence-only inputs, it can run AlphaFold2 on the fly to produce a structure for structure-aware inference.
4. Under the hood, xBind exposes two independently trained frameworks: EquiPPIS for protein–protein interfaces and EquiPNAS for protein–DNA/RNA interfaces, both designed to be robust to imperfect structures (including AlphaFold models).
5. Benchmarking emphasizes performance on AlphaFold2-predicted structures, reflecting real-world usage where experimental complexes are missing. Reported comparisons show statistically significant gains (95% confidence, P < 0.05) over strong baselines using ROC-AUC and PR-AUC across multiple test sets.
6. Example performance (AlphaFold inputs): for protein–protein, EquiPPIS improves over GraphPPIS on Test_60 (ROC-AUC 0.7912 vs 0.7667; PR-AUC 0.4516 vs 0.3956). For protein–DNA, EquiPNAS leads on Test_181 (ROC-AUC 0.9159; PR-AUC 0.3717) and Test_129 (ROC-AUC 0.9387; PR-AUC 0.569). For protein–RNA, EquiPNAS improves over GraphBind on Test_117 (ROC-AUC 0.8856 vs 0.7942; PR-AUC 0.3118 vs 0.2019).
7. The web UI is built for interpretability and calibration: it provides interactive sequence views, 3D visualization of predicted interface residues, per-residue likelihood plots, and user-adjustable probability thresholds to tune precision/recall for different downstream needs.
8. Output artifacts are designed for reuse: downloadable structure used for prediction, residue-level probability files, and a packaged archive with complete outputs and run metadata; jobs can be public or private, with optional email notification.
9. The paper also outlines near-term directions: calibrating predictions using residue-wise uncertainty signals (e.g., experimental B-factors or AlphaFold pLDDT) and incorporating conformational dynamics/multistate behavior to better model binding under structural heterogeneity.
📜Paper: doi.org/10.1093/nar/gkag425
#Bioinformatics #ComputationalBiology #ProteinScience #StructuralBioinformatics #DeepLearning #GraphNeuralNetworks #ProteinLanguageModels #ESM2 #AlphaFold #ProteinInteractions #NARWebServerIssue
1
3
37
2,274
mTM-align2: A Server for Real-time Protein Structure Database Search and Alignment
1. mTM-align2 is presented as a real-time web server that searches structurally similar proteins across ~3 million structures (experimental predicted) in seconds, while supporting both monomeric proteins and multimeric complexes.
2. The key idea is to replace expensive all-vs-all structural alignments with fast cosine-similarity search over precomputed structural fingerprints (embeddings), then optionally run detailed pairwise/multiple alignments with mTM-align for interpretability and validation.
3. For monomers, the server encodes a query using residue-level embeddings from the pretrained inverse-folding protein language model ESM-IF, aggregates them into a protein-level vector (sum pooling), and uses contrastive learning so cosine similarity correlates with structural similarity.
4. For multimers, it combines (a) chain-wise monomer embeddings and (b) a rotation-invariant global shape descriptor based on 3D Zernike moments; the final similarity (Q-score) is a weighted sum of chain similarity (IF-score) and shape similarity (ZP-score), with weights α=1 and β=0.3.
5. mTM-align2 integrates multiple major databases in one interface: PDB (including large monomer and multimer sets), SCOPe and CATH domain databases, BFVD viral structures, plus several AlphaFold DB subsets (Swiss-Prot, organism set, global health set), enabling cross-database retrieval rather than siloed searches.
6. The server offers two search modes: a high-accuracy mode that adds a fast TM-align-based filtering step to improve precision, and a high-speed mode that skips filtering for near-instant results; results are returned as ranked hits (top 1000) and can be emailed as CSV for batch workflows.
7. Beyond retrieval, the output workflow is designed for downstream analysis: users can launch pairwise superposition with TM-align metrics (TM-score, RMSD) and residue-level correspondence, or run multiple structure alignment (up to 10 selected hits via the UI) to identify conserved cores and generate a structure-based phylogenetic tree.
8. A practical annotation feature is included for PDB hits: after superposition, the server transfers ligand context from Q-BioLiP and marks query residues within 5 Å of the aligned ligand as putative binding residues, providing fast template-based binding-site hints (with the paper noting it is not meant to replace specialized binding-site predictors).
9. Benchmarks against Foldseek variants suggest complementary strengths for monomer search (mTM-align2 wins on more test cases by summed true-positive TM-scores in top-100, while Foldseek leads on others), and stronger multimer retrieval performance versus foldseek-multimer (top-50 recall ~82.4% vs 77.7%; precision ~35.08% vs 27.66%).
10. A case study on the MscL mechanosensitive channel highlights sensitivity to remote homologs and conformational diversity: against AFDB-SwissProt, mTM-align2 retrieves many more true positives than Foldseek, and multiple alignment of top hits reveals strictly conserved transmembrane helices linked to gating architecture.
💻Code: ngdc.cncb.ac.cn/biocode/tool…
📜Paper: doi.org/10.1093/gpbjnl/qzag0…
#ProteinStructure #StructuralBioinformatics #AlphaFold #ProteinComplexes #StructureSearch #BioinformaticsTools #ESM #ContrastiveLearning #ZernikeMoments #TMAlign

5
39
2,638
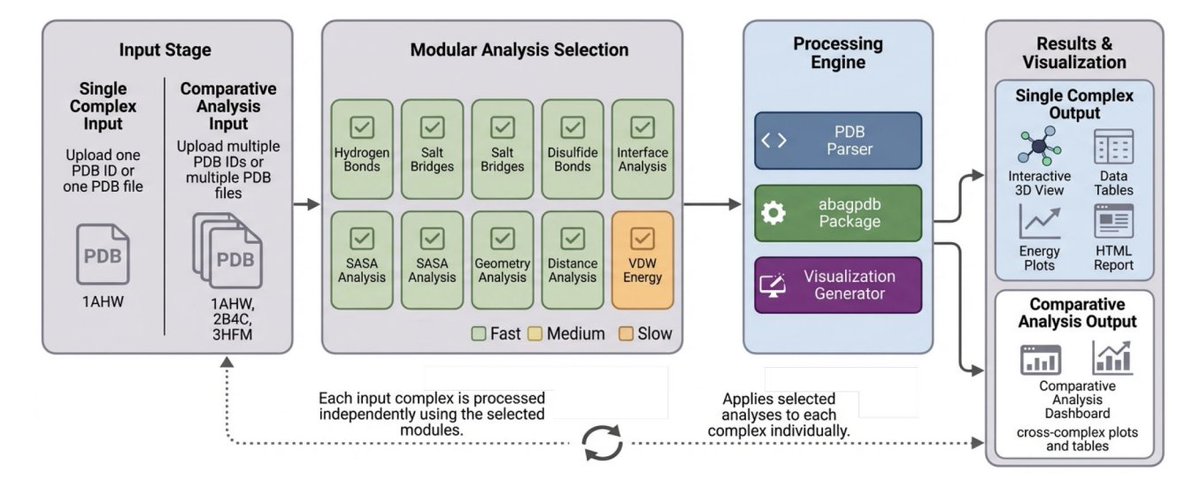
Comparative Structural Analysis of Protein Complexes With SPICE
1 SPICE (Structural Protein Interaction Complex Evaluator) is a web-based platform for rapid, customizable structural analysis of protein complexes directly from PDB structures, targeting common bottlenecks in interface characterization and variant comparison.
2 The core differentiator is modular workflow design: users select only the analyses they need (checkbox-style), which the authors report can reduce typical turnaround from minutes to seconds while keeping the workflow reproducible and easy to rerun.
3 SPICE emphasizes comparative, multi-complex analysis as a first-class feature: it can evaluate wild-type vs mutant complexes, different binding partners, or homologs, then aggregate results into differential tables/plots rather than requiring manual post-processing across tools.
4 The platform covers interface interaction detection (hydrogen bonds, salt bridges, disulfide bonds) with explicit geometric criteria, enabling consistent, residue-level interaction accounting across complexes and across variants.
5 Interface mapping is residue-centric: residues within a proximity threshold (typically 5.0 Å) are labeled as interface, with per-residue contact frequency and contact-type breakdowns to help identify hotspots and compare how specific positions change across variants.
6 Energetics and accessibility modules include SASA/BSA (via FreeSASA; Lee–Richards with 1.4 Å probe), RSA-based residue classification, and optional per-residue van der Waals interaction estimates (Lennard–Jones 6–12) to highlight favorable packing vs potential steric clashes (relative rather than absolute energies).
7 Geometry/quality assessment is integrated into the same workflow: Ramachandran plots (with interface residues highlighted), side-chain chi/rotamer analysis, bend angles for backbone curvature, and interactive inter-residue distance heatmaps with tunable cutoffs.
8 For multi-complex comparisons, SPICE supports synchronized side-by-side 3D inspection (optional alignment via Biopython for visualization), and mutation impact summaries such as ΔVDW vs ΔSASA scatter plots to separate energetic vs geometric drivers of binding changes.
9 Case studies illustrate (i) antibody D1.3–lysozyme (1A2Y) for single-complex interface discovery, contact profiling, distance matrices, and per-residue burial/VDW mapping; and (ii) PD1–pembrolizumab (5GGS) vs a single-mutation variant to localize mutation-driven shifts in burial, contacts, bond types, and energetics with coordinated plots 3D exploration.
💻Code: github.com/fbabd/PyPdbComple…
📜Paper: doi.org/10.1093/nar/gkag415
#StructuralBioinformatics #ProteinProteinInteractions #ProteinComplexes #WebServer #AntibodyEngineering #DrugDesign #ComputationalBiology #PDB #BioinformaticsTools
1
7
997
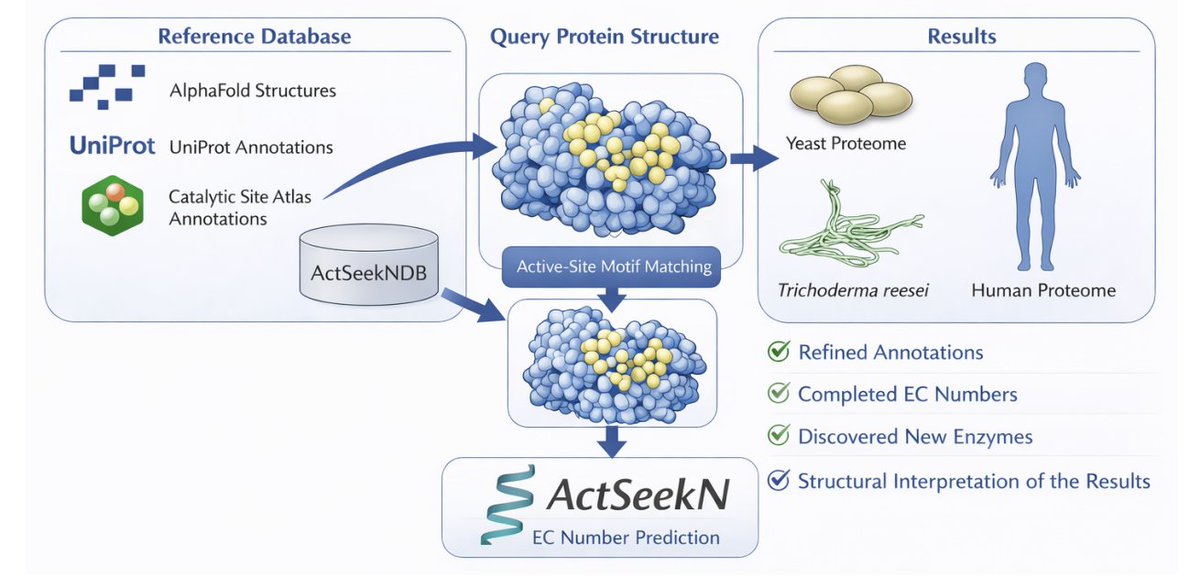
ActSeekN: A Structural-Motif–Based Pipeline for Interpretable Enzyme Function Annotation
1. ActSeekN annotates enzyme function by matching local 3D catalytic/binding-site geometry (structural motifs) rather than relying on global sequence similarity, aiming to work better for low-identity proteins and convergent evolution while keeping predictions mechanistically interpretable.
2. The key enabling piece is ActSeekNDB: a large reference database built from AlphaFold-predicted structures UniProt EC annotations curated catalytic residue information (including CSA). It contains 39,761 proteins spanning 6,050 EC numbers, addressing the historical bottleneck of small curated active-site databases.
3. Database construction workflow: for each UniProt EC number, up to 250 proteins were collected and structurally clustered with TM-align; clusters were defined using an RMSD < 0.3 threshold, then one representative structure per cluster was selected to represent that EC-linked structural family.
4. Active/binding-site residues in ActSeekNDB are assigned by intersecting UniProt-annotated residues across proteins in a structural cluster and combining them with structurally conserved residues from alignments; CSA entries are integrated and missing residues can be supplemented by conserved residues when CSA provides fewer than three.
5. When UniProt residue-level annotations are missing, an AI-based agent (described as based on ChatGPT5.1) assists by selecting three spatially clustered residues, guided by coordinates, conserved residues, EC context, and prior curated annotations—explicitly to propose plausible catalytic-site candidates for motif seeding.
6. The ActSeekN tool combines the previously published ActSeek active-site search with ActSeekNDB, implemented as a standalone C program with multithreading, KD-tree nearest-neighbor search, and optional CUDA GPU acceleration; reported average runtime is ~15 seconds per protein on the Price dataset.
7. Benchmarking used the same datasets and evaluation procedure as CLEAN-Contact (New392 and Price), keeping the top five EC predictions and filtering hits below 0.15% residue position similarity; ActSeekN reported higher Precision/Recall/F1 than CLEAN-Contact on both datasets, while also returning the specific residues supporting each annotation.
8. Proteome-scale applications: in Saccharomyces cerevisiae, ActSeekN reported 88.9% overall prediction accuracy, completed 192 previously incomplete EC numbers, and proposed EC annotations for proteins lacking ECs but having GO molecular-function terms, often yielding consistent enzyme activities (examples include ATPases, kinases, lyases, hydrolases).
9. Case studies highlight how residue-level interpretability is used to reconcile disagreements: yeast HMF1 (P40037) is annotated as a growth inhibitor but is predicted as EC 3.5.99.10 based on identical catalytic residues and strong fold match; an uncharacterized membrane protein (P42840) is predicted as a palmitoyl-protein thioesterase (EC 3.1.2.22) with matching key active-site residues despite imperfect global alignment.
10. In humans, ~90% of predictions matched UniProt EC assignments; ActSeekN refined 521 incomplete EC annotations and proposed plausible enzymatic functions for proteins with broad/non-enzymatic descriptions. The paper also emphasizes limitations: conserved motifs can persist after functional divergence (e.g., SLC3A1 predicted as a hydrolase-like scaffold), so results are positioned as mechanistically grounded hypotheses rather than definitive in vivo proof.
💻Code: github.com/Aalto-Microbial-P…
📜Paper: biorxiv.org/content/10.64898…
#computationalbiology #bioinformatics #enzymes #proteinstructure #alphafold #functionalannotation #structuralbioinformatics #proteomics #metabolicengineering #biotechnology
4
18
1,744
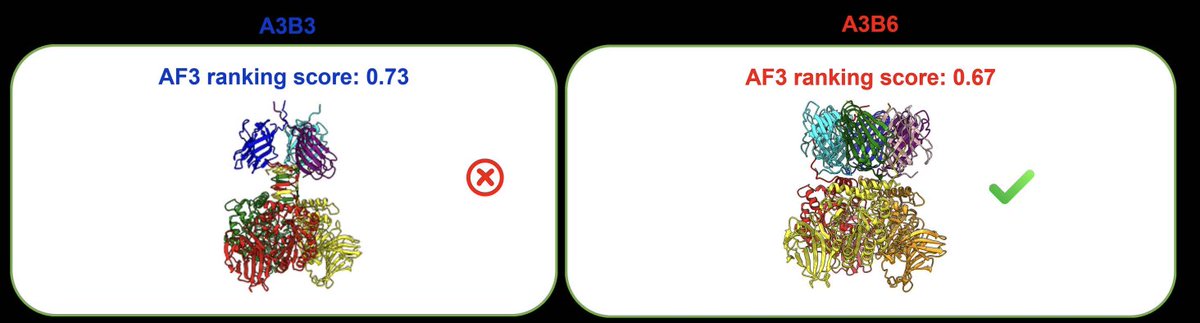
PreStoi allows accurate prediction of protein complex stoichiometry by integrating AlphaFold3 and template information
1. The paper addresses a practical bottleneck in multimer structure prediction: AlphaFold-style complex modeling typically assumes stoichiometry is known, but many real complexes lack reliable copy-number annotations. PreStoi targets the upstream task: predicting how many copies of each subunit are present.
2. Core idea: treat stoichiometry as a discrete search-and-rank problem. PreStoi (i) proposes plausible stoichiometry candidates, (ii) builds AlphaFold3 models for each candidate, (iii) ranks candidates using AlphaFold3 ranking scores, and (iv) refines decisions using homologous complex-template evidence when available.
3. CASP16 Phase 0 (blind stoichiometry) results: using the MULTICOM_AI predictor with PreStoi, the correct stoichiometry was ranked top-1 for 71.4% of targets (20/28) and appeared in the top-3 for 92.9% (26/28). Compared with other CASP16 approaches, performance was strongest when considering both top-1 accuracy and “any-submitted” accuracy.
4. The integration matters: AlphaFold3-only ranking was substantially weaker than the combined system. On CASP16 targets, AF-max (max AF3 ranking score) achieved 50% top-1, AF-avg (average score) 42.9%, and template-based prediction alone 50%; the integrated decision rule reached 71.4% top-1.
5. Why templates help: when significant complex templates exist, template-based stoichiometry is often more precise for top-1 selection (82.4% accuracy on applicable CASP16 targets), and it can override AF3 ranking mistakes. Templates also cover cases where AF3 cannot be run (e.g., very large complexes exceeding the AlphaFold3 web-server 5000-residue limit).
6. Why AlphaFold3 helps: templates are missing for a substantial fraction of targets (about 39% in CASP16). In those cases, AF3 ranking provides a workable signal to narrow down candidates, and AF-max/AF-avg can still succeed without a full complex template spanning all subunits.
7. A key failure mode is “compatible stoichiometries” that yield highly similar structures, especially hetero-dimer (A1B1) vs hetero-tetramer (A2B2). The models can be partially/fully superimposable, making AF3 ranking scores (and sometimes template heuristics) struggle to confidently separate them.
8. Additional hard cases highlight missing priors beyond AF3 ranking scores: (i) hierarchical oligomerization (subunits form homo-oligomers first, then assemble), (ii) symmetry considerations (symmetric stoichiometries may be more plausible than asymmetric high-scoring ones), and (iii) filaments with large or variable copy numbers (e.g., A9B18), where candidate enumeration becomes intractable without strong external constraints.
9. Large-scale benchmark (P16-BMS; 2,014 post-CASP16 “blind” PDB complexes): automated template evidence alone reached 46.2% top-1 (64.9% top-3). For hetero-multimers, using AlphaFold3 to re-rank the top-10 template-derived candidates improved top-1 into ~46–49% range, illustrating complementarity between template evidence (candidate generation/prior) and AF3 scores (re-ranking signal).
10. Method details of the automated pipeline: templates are found per subunit via MULTICOM4 (Jackhmmer/MSA, HHsearch). Candidate copy numbers are supported and weighted by −log10(E-value). A multi-subunit template promotion step boosts candidates consistent with partial/full complex templates spanning multiple subunits, while filtering out candidates exceeding the 5000-residue AF3 feasibility limit.
💻Code: github.com/jianlin-cheng/pre…
📜Paper: doi.org/10.1038/s42003-026-1…
#ProteinComplexes #Stoichiometry #AlphaFold3 #CASP16 #StructuralBioinformatics #ComputationalBiology #ProteinStructure #MultimerPrediction #Templates #Benchmarking
5
25
2,292
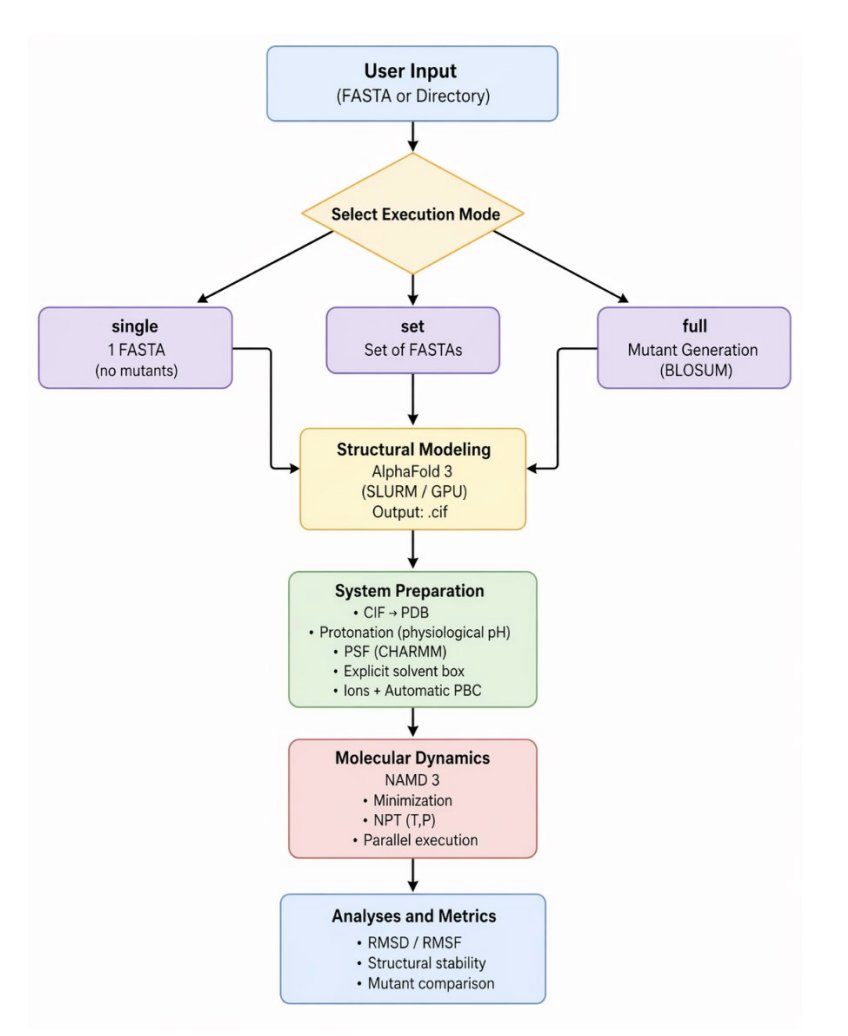
AlphaUnfold: Probing Potential Unfolding and Structural Fragility in AlphaFold3 Models via Short-Time High-Pressure MD
1 AlphaUnfold is an automated “stress-test” pipeline that takes AlphaFold3 (AF3) models and subjects them to short (5 ns) high-pressure MD (1000 atm) in NAMD3, aiming to reveal structural fragility and early unfolding signals that static confidence scores can miss.
2 The key result is a strong inverse relationship between AF3 average pLDDT and MD structural drift: after 5 ns high-pressure MD, lower-confidence models show markedly higher RMSD. The reported regression is RMSD = 35 − 0.34·pLDDT (R² = 0.78, p < 0.001), implying ~3.4 Å less RMSD per 10 pLDDT points.
3 A central validation claim is that 5 ns at 1000 atm captures similar “where it moves” patterns as much longer conventional MD: the study compares high-pressure 5 ns runs to standard-pressure trajectories (notably 200 ns at 1 atm) and finds consistent ranking/patterns of structural drift, with high-pressure runs converging quickly to metastable states (lower RMSD standard error in the final segment).
4 Beyond global RMSD, AlphaUnfold emphasizes local vulnerability mapping: per-residue RMSF profiles align with local pLDDT, where low pLDDT regions (e.g., <50) tend to show high fluctuations (e.g., >5 Å), while high pLDDT regions (>90) remain comparatively rigid (often <2 Å RMSF). This is presented as a practical way to pinpoint metastable segments for redesign.
5 The workflow is designed for throughput and reproducibility: AF3 prediction and pLDDT extraction, automated MD system preparation (solvation/ions/minimization), 5 ns high-pressure production in triplicate, then automated reporting of RMSD/RMSF plus compactness/exposure metrics (RoG, SASA), with optional shape/contact analyses (ellipsoid index, COCaDA contacts).
6 The paper frames the pipeline as particularly useful for protein engineering in single-cell protein (SCP) contexts: when enriching essential amino acids (e.g., via BLOSUM-guided substitutions), AlphaUnfold can serve as a rapid computational filter to reject designs that look plausible in AF3 but are dynamically fragile under stress.
7 The authors highlight important outliers where pLDDT may not predict MD behavior, illustrating how missing chemistry in simulation setup can dominate outcomes: a cited case (Q96M98-2) has high average pLDDT (~85) but unusually high RMSD (11.7 Å), potentially due to omitted stabilizers such as explicitly defined disulfide bonds, or missing cofactors/ligands/ions.
8 The broader message is methodological: pLDDT is informative but static; coupling AF3 with a brief, physics-based perturbation (high-pressure MD) can provide an “experimental-like” robustness check and actionable localization of weak spots, intended to improve confidence in downstream use of AI-generated structures.
💻Code: github.com/pegados/pipeline_…
📜Paper: biorxiv.org/content/10.64898…
#AlphaFold3 #MolecularDynamics #ProteinStructure #ComputationalBiology #ProteinEngineering #NAMD #StructuralBioinformatics #HighPressureMD #SCP
3
7
50
2,945
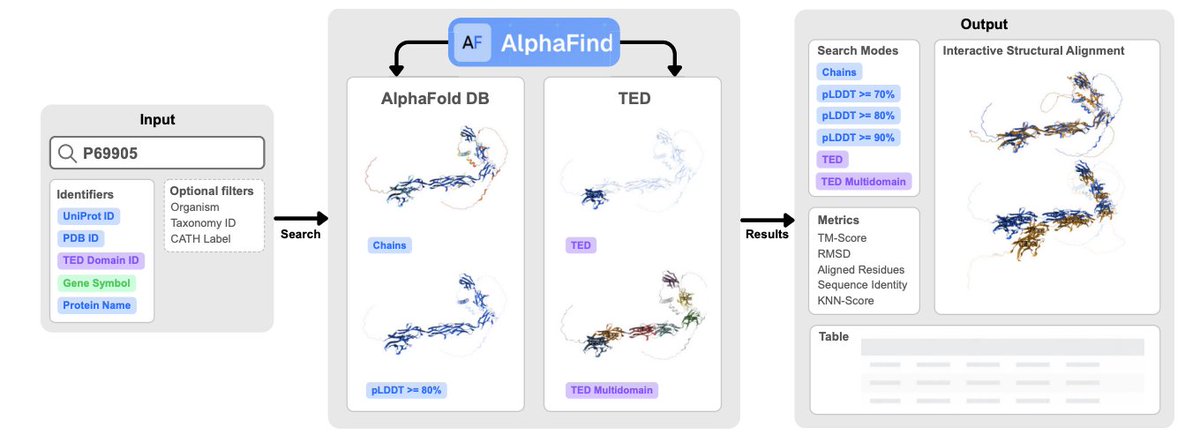
AlphaFind v2: Similarity search in AlphaFold DB and TED domains across structural contexts
1 AlphaFind v2 is a web server for fast, structure-based similarity search at AlphaFold DB scale, combining embedding-based prefiltering with alignment-based refinement to keep results biologically interpretable (TM-score/RMSD) while staying interactive.
2 The key design idea is “search across structural contexts”: users can search full chains, restrict comparisons to high-confidence regions using AlphaFold pLDDT thresholds (70/80/90), search TED domains, or run a TED Multidomain mode that captures domain combinations rather than single-domain matches.
3 The workflow is staged for responsiveness: Phase 2 returns immediate approximate kNN results from a vector database (top k=100 by cosine similarity), while Phase 3 runs asynchronously in the background to refine rankings using US-align and report TM-score, RMSD, aligned residues, and interactive superpositions.
4 pLDDT-aware search directly addresses a common AlphaFold-era problem: low-confidence/disordered regions can dominate alignments and hide true homologs. By trimming residues below chosen pLDDT thresholds, AlphaFind v2 focuses similarity on stable structural cores.
5 Domain-level search is integrated via TED: AlphaFind v2 supports direct TED domain retrieval and alignment restricted to domain residue boundaries, enabling more fine-grained detection of shared folds when full-length proteins differ in architecture.
6 TED Multidomain mode targets proteins where function/evolution is encoded in domain composition and order. It aggregates multiple domain-to-domain matches into a single score/alignment, aiming to recover “same architecture” relationships that single-domain hits would miss.
7 A distinctive interface feature in TED Multidomain is interactive weighting: sliders adjust each matched domain pair’s contribution, updating the 3D alignment view to move between (i) inspecting one domain precisely and (ii) assessing global multi-domain arrangement.
8 Under the hood, AlphaFold DB v4 chains are embedded into 1536D vectors using an ESM3-based pipeline; additional embeddings are computed after removing low-confidence residues (pLDDT < 70/80/90). TED domains use precomputed 128D Foldclass embeddings.
9 Engineering choices focus on scalable, low-latency search: OpenSearch vector DB with HNSW (16x compression, on-disk), a Python/Flask REST API, Celery Redis for async refinement jobs, PostgreSQL for state/caching, and Kubernetes for horizontal scaling.
10 Reported benchmarks show rapid retrieval plus strong refinement quality: approximate results in ~2.4 s for chains and ~0.49 s for domains, with refinement completing in tens of seconds; evaluation indicates higher average TM-scores than AlphaFind v1, FoldSeek server (TM computed separately), and Merizo-search (domains), with statistical significance (P < 0.05).
11 Case study (PIN3 auxin carrier): full-chain search struggles due to large disordered loops, but pLDDT ≥ 90 mode finds homologs with TM-score up to 0.947, illustrating how confidence-filtered structural search can recover relationships obscured by structural “noise”.
12 Case study (NCAM1): TED Multidomain mode captures the characteristic Ig-domain fibronectin type III arrangement, helping identify proteins with similar multidomain architecture; interactive reweighting helps resolve cases where domain positions differ across predictions.
📜Paper: doi.org/10.1093/nar/gkag372
#ProteinStructure #AlphaFold #StructuralBioinformatics #ProteinDomains #SimilaritySearch #Embeddings #WebServer #TMscore #CATH #TED #ComputationalBiology
1
6
41
2,635
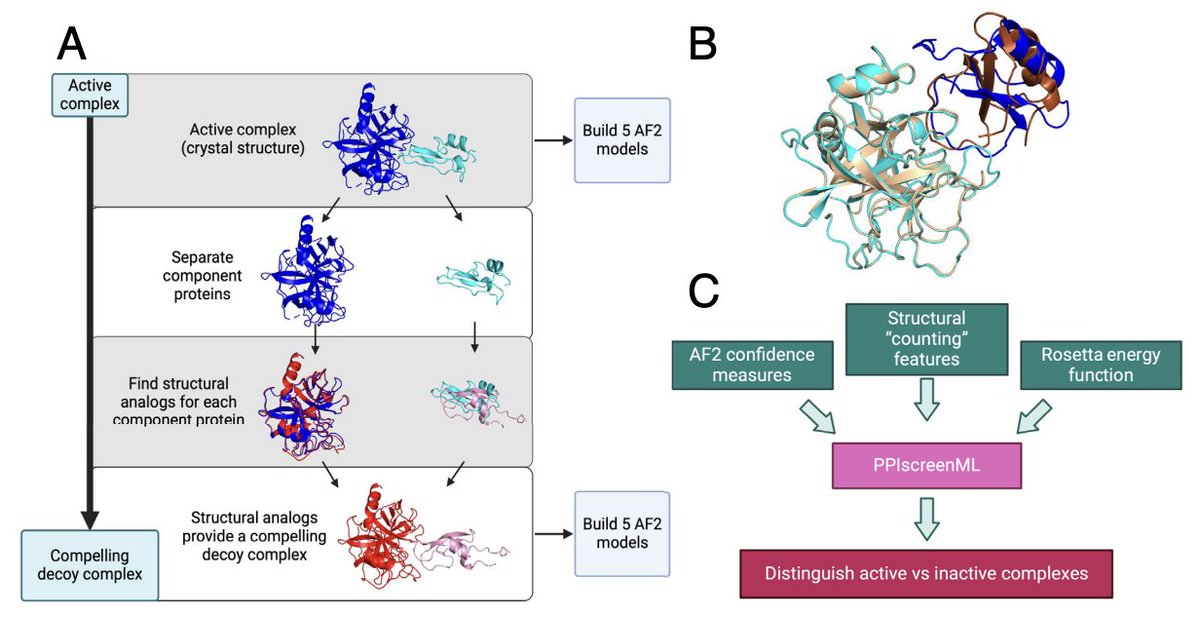
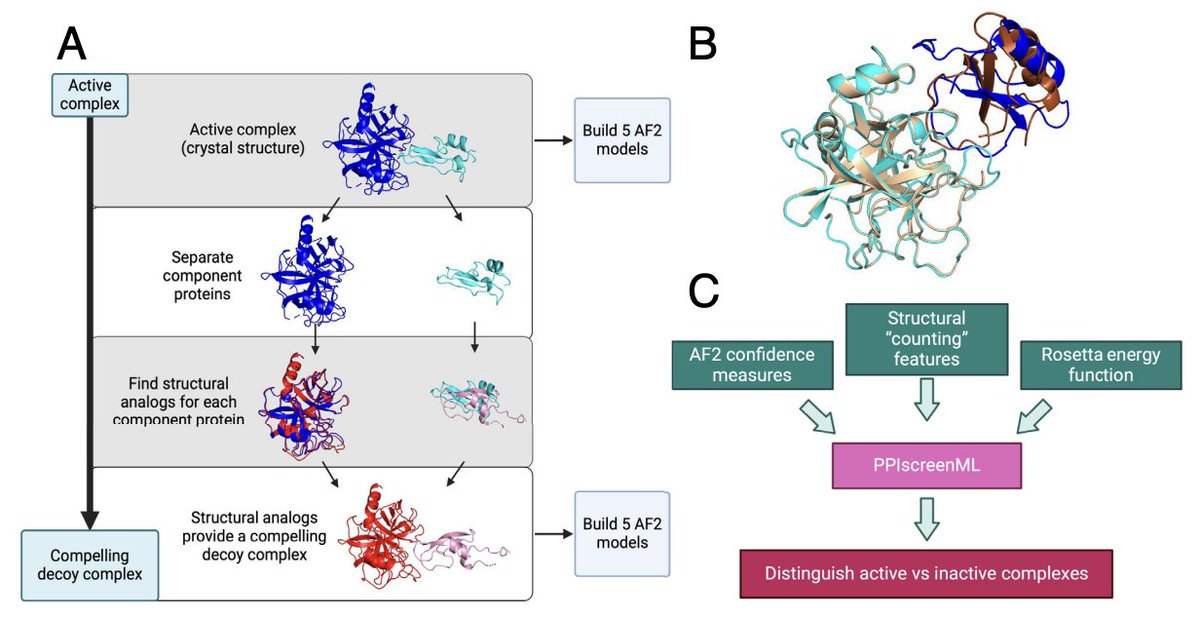
PPIscreenML is a method for structure-based screening of protein-protein interactions using AlphaFold
1. Mischley et al. present PPIscreenML, a structure-based ML classifier that predicts whether a candidate protein pair truly interacts, using AlphaFold2-Multimer models as input rather than relying on sequence-only inference.
2. The key idea is to train explicitly on the interaction-vs-noninteraction task (not just “model quality”): PPIscreenML learns to separate AF2 models of real interacting heterodimers from AF2 models of “compelling decoys” that look structurally plausible.
3. Decoy generation is a central innovation: for each true PDB heterodimer, each partner is replaced by its closest structural analog (by TM-score) from a nonredundant set, then aligned into the original complex geometry—creating inactive pairs that mimic the geometry of true interfaces and are hard to dismiss by trivial heuristics.
4. Dataset scale and realism: 1481 nonredundant heterodimeric PDB complexes (<=30% sequence identity; excluding homodimers and antibody/antigen). Five AF2-Multimer v2.3 predictions per active and per decoy. Training includes only AF2 active models with DockQ >= 0.23 (to avoid learning from mis-docked “actives”), but the held-out test set keeps mis-docked actives to better reflect prospective screening.
5. Feature design blends AF2 confidence with physics-inspired energetics: 57 features total, spanning AF2 confidence metrics (pLDDT/pTM/PAE-derived terms), structural “counting” descriptors of interfaces, and Rosetta energy terms computed on the predicted complexes.
6. Model selection: several standard classifiers perform similarly, with gradient-boosted trees (XGBoost) best overall. On a completely held-out test set, the full feature model reaches ROC-AUC ~0.892 when scoring each candidate by the best of 5 AF2 models (a practical screening strategy).
7. A compact 7-feature version retains nearly the same performance (test ROC-AUC ~0.884), suggesting much of the signal is captured by a small set of interpretable interface cues: an interface-PAE statistic, interface charge count, and multiple Rosetta interfacial terms (LJ attractive/repulsive, solvation, electrostatics) plus a beta-sheet-related interface fraction.
8. Benchmark vs commonly used AF2-derived scores: on the same held-out test set, PPIscreenML outperforms iPTM and pDockQ for classifying interacting vs noninteracting pairs (AUC ~0.884 vs ~0.843 for iPTM and ~0.710 for pDockQ), highlighting the benefit of training specifically for screening rather than for structure-quality assessment.
9. Generalization test in a difficult “structurally conserved but selective” regime: across the TNF superfamily (18 ligands x 28 receptors = 504 pairs; only 36 known binders), AF2 can model many pairs in similar poses regardless of true binding. PPIscreenML nonetheless recapitulates specificity well (ROC-AUC ~0.93), with the top-scoring receptor matching a true interactor for 14/18 ligands (and within top-2 for 17/18), despite TNFSF not being in training (and training restricted to dimers).
💻Code: github.com/victoria-mischley…
📜Paper: doi.org/10.7554/eLife.98179
#ProteinProteinInteractions #AlphaFold #AlphaFoldMultimer #ComputationalBiology #StructuralBioinformatics #MachineLearning #Rosetta #Interactome #SystemsBiology #DrugDiscovery
1
2
25
11,363
PPIscreenML is a method for structure-based screening of protein-protein interactions using AlphaFold
1. Mischley et al. present PPIscreenML, a structure-based ML classifier that predicts whether a candidate protein pair truly interacts, using AlphaFold2-Multimer models as input rather than relying on sequence-only inference.
2. The key idea is to train explicitly on the interaction-vs-noninteraction task (not just “model quality”): PPIscreenML learns to separate AF2 models of real interacting heterodimers from AF2 models of “compelling decoys” that look structurally plausible.
3. Decoy generation is a central innovation: for each true PDB heterodimer, each partner is replaced by its closest structural analog (by TM-score) from a nonredundant set, then aligned into the original complex geometry—creating inactive pairs that mimic the geometry of true interfaces and are hard to dismiss by trivial heuristics.
4. Dataset scale and realism: 1481 nonredundant heterodimeric PDB complexes (<=30% sequence identity; excluding homodimers and antibody/antigen). Five AF2-Multimer v2.3 predictions per active and per decoy. Training includes only AF2 active models with DockQ >= 0.23 (to avoid learning from mis-docked “actives”), but the held-out test set keeps mis-docked actives to better reflect prospective screening.
5. Feature design blends AF2 confidence with physics-inspired energetics: 57 features total, spanning AF2 confidence metrics (pLDDT/pTM/PAE-derived terms), structural “counting” descriptors of interfaces, and Rosetta energy terms computed on the predicted complexes.
6. Model selection: several standard classifiers perform similarly, with gradient-boosted trees (XGBoost) best overall. On a completely held-out test set, the full feature model reaches ROC-AUC ~0.892 when scoring each candidate by the best of 5 AF2 models (a practical screening strategy).
7. A compact 7-feature version retains nearly the same performance (test ROC-AUC ~0.884), suggesting much of the signal is captured by a small set of interpretable interface cues: an interface-PAE statistic, interface charge count, and multiple Rosetta interfacial terms (LJ attractive/repulsive, solvation, electrostatics) plus a beta-sheet-related interface fraction.
8. Benchmark vs commonly used AF2-derived scores: on the same held-out test set, PPIscreenML outperforms iPTM and pDockQ for classifying interacting vs noninteracting pairs (AUC ~0.884 vs ~0.843 for iPTM and ~0.710 for pDockQ), highlighting the benefit of training specifically for screening rather than for structure-quality assessment.
9. Generalization test in a difficult “structurally conserved but selective” regime: across the TNF superfamily (18 ligands x 28 receptors = 504 pairs; only 36 known binders), AF2 can model many pairs in similar poses regardless of true binding. PPIscreenML nonetheless recapitulates specificity well (ROC-AUC ~0.93), with the top-scoring receptor matching a true interactor for 14/18 ligands (and within top-2 for 17/18), despite TNFSF not being in training (and training restricted to dimers).
💻Code: github.com/victoria-mischley…
📜Paper: doi.org/10.7554/eLife.98179
#ProteinProteinInteractions #AlphaFold #AlphaFoldMultimer #ComputationalBiology #StructuralBioinformatics #MachineLearning #Rosetta #Interactome #SystemsBiology #DrugDiscovery
13
913
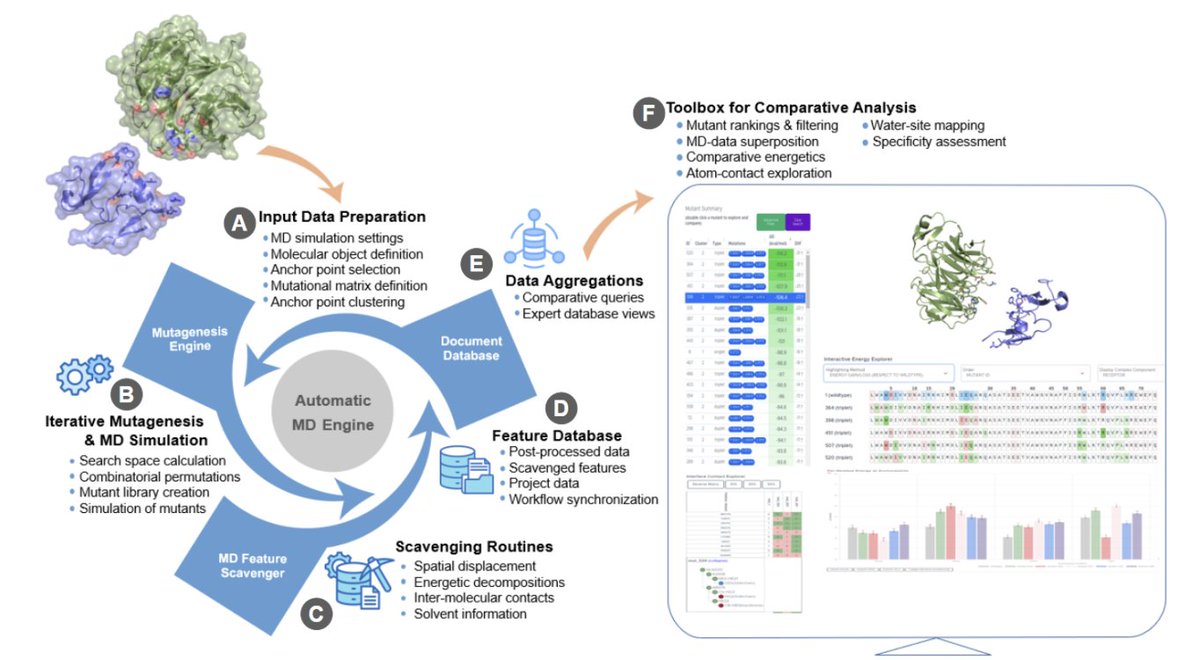
DyME: An MD-based engine exploiting HTP mutagenesis for protein engineering and recognition mimicry
1. DyME (Dynamic Mutagenesis Engine) is a distributed, MD-first platform designed to run and compare up to thousands of mutant complexes, turning what is usually a fragmented “mutate → simulate → analyze” workflow into a single automated pipeline for protein recognition mimicry (PRM).
2. The core idea is systematic, combinatorial interfacial mutagenesis (single/double/triple variants) coupled to solvated MD, with all trajectory-derived features scavenged into a central database so large mutant libraries can be queried and compared without manual per-simulation handling.
3. Input is any 3D structure of a protein-protein, protein-peptide, or protein-DNA complex. Users define “molecular objects” and choose one as the mutable ligand/partner; DyME proposes interfacial “anchor points” from contact analysis, which users can refine and cluster for combinatorial library design.
4. A key innovation is the data-management and analysis layer: DyME stores extracted MD features (e.g., RMSD, ΔG estimates, per-residue decomposition, pairwise contact frequencies, solvent features) in MongoDB, enabling fast aggregation queries across millions of records spanning many simulations.
5. DyME’s “anchor point clustering” controls combinatorial explosion by organizing positions into clusters and enumerating non-redundant singlet/doublet/triplet permutations per cluster, while the GUI provides estimated mutant counts and projected GPU-hours during project setup.
6. The distributed backend follows a producer-consumer model: MD worker nodes poll the database for pending mutants, generate mutant structures (Modeller), build Amber-format systems (tleap), and run OpenMM simulations on idle GPUs; scavenger nodes then post-process trajectories (AmberTools/CPPTRAJ, MDTraj, MDAnalysis, MMPBSA.py) and push curated features back to the database.
7. DyME emphasizes recognition beyond direct contacts by integrating interfacial water analysis: the Toolbox for Comparative Analysis (TCA) includes a Water Site Explorer that maps water-sites from trajectories and summarizes water-mediated contacts and residence, supporting mechanistic interpretation of solvent-driven recognition changes across mutants.
8. The interactive web-based TCA is designed for comparative exploration at scale: Mutant Explorer ranks mutants by binding energy metrics and supports multi-mutant overlay of structures, RMSD traces, per-anchor-point energy bars, sequence-position energy “heat” views, pairwise contact/energy maps, and contact tables—reducing the need to manually inspect large decomposition outputs.
9. DyME also targets specificity engineering with a dedicated “Specificity Finder” that compares two projects sharing the same mutable ligand but different receptors, identifying mutations that increase affinity for one receptor while decreasing it for the other using database aggregations across matched mutants.
10. Case study: DyME is validated on SH3 recognition mimicry using the classic Abl-SH3 vs Fyn-SH3 system and the 3bp-1 peptide mutational landscape. The platform reproduces trends consistent with experimental affinity/specificity shifts (via ΔG vs reported Kd correlation) and recovers experimentally described water-mediated interactions at SH3 interfaces, illustrating how DyME supports affinity and selectivity design rationales.
💻Code: github.com/pisabarro-group/D…
📜Paper: biorxiv.org/content/10.64898…
#computationalbiology #moleculardynamics #proteinengineering #structuralbioinformatics #mutagenesis #proteinproteininteractions #proteinDNA #OpenMM #Amber #FAIRdata
2
1
19
2,143
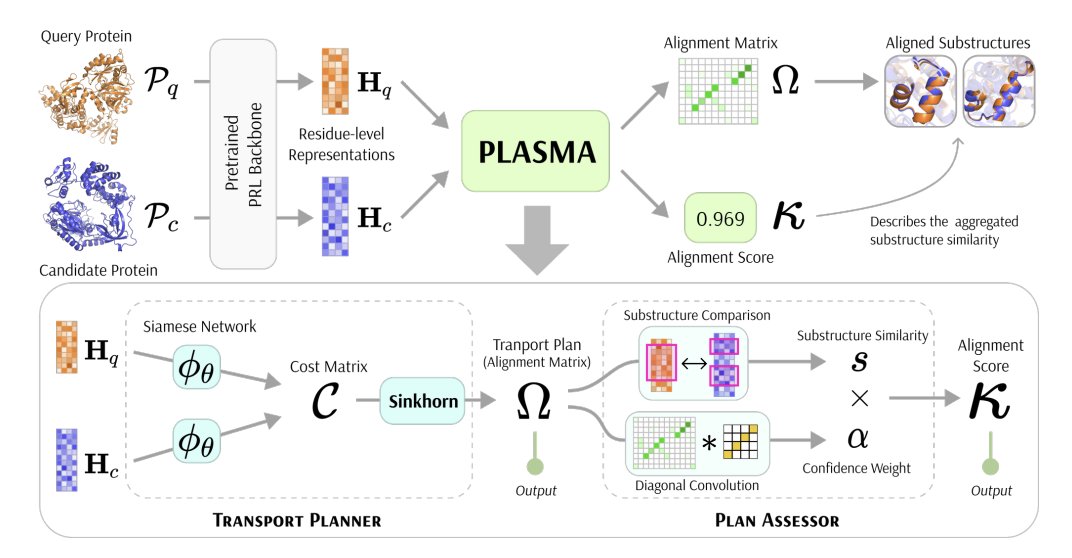
Fast and Interpretable Protein Substructure Alignment via Optimal Transport
1 PLASMA frames residue-level local structural alignment as an entropy-regularized optimal transport (OT) problem, producing a soft alignment matrix (residue–residue correspondences) plus a normalized similarity score in [0,1] for each protein pair.
2 Core design: a Transport Planner learns a residue-pair cost matrix (siamese network over residue embeddings hinge nonlinearity) and solves OT with differentiable Sinkhorn iterations, enabling GPU-parallel, end-to-end trainable local alignment.
3 Unlike global structure alignment or dynamic-programming local methods, the OT formulation naturally supports partial, variable-length, and non-sequential motif matches—important when functional residues cluster in 3D but are far apart in sequence or embedded in different folds.
4 A key interpretability point is that PLASMA outputs an explicit alignment matrix Ω with probabilistic meaning (transport weights), rather than only a best-path alignment or an unbounded heuristic score; this makes it easier to inspect which residues drive the match.
5 To avoid biologically meaningless “forced” full matching typical of doubly-stochastic Sinkhorn, PLASMA uses implicit partial alignment via (i) early stopping of Sinkhorn iterations to preserve sparsity and (ii) a temperature parameter controlling alignment sharpness.
6 The Plan Assessor converts Ω into a single score: it computes substructure similarity as cosine similarity between summed embeddings of matched residues, then applies a confidence weight derived from a diagonal “identity-kernel” convolution over Ω to reward contiguous diagonal segments (coherent local alignments) and downweight fragmented matches.
7 Training uses two objectives: (i) a binary classification loss on whether a pair contains a matched functional substructure, and (ii) a Label Match Loss that only supervises annotated residues to avoid penalizing unlabeled-but-valid correspondences—important because functional annotations can be incomplete.
8 A practical contribution is PLASMA-PF, a training-free parameter-free variant that skips the siamese cost network and operates directly on residue embeddings, providing a usable option when labeled training data are unavailable.
9 On the VenusX residue-level functional alignment benchmark (motifs, binding sites, active sites) with <50% sequence identity and an extrapolation split to unseen InterPro families, PLASMA reports strong detection and localization performance (e.g., ROC-AUC often ~0.97–0.99) and improves alignment localization (LMS) over the training-free variant.
10 Efficiency: reported inference is ~10 ms per protein pair for PLASMA (~7 ms for PLASMA-PF), roughly ~50× faster than superposition-based global structure alignment (TM-align/Foldseek) and ~3× faster than a dynamic-programming embedding-based local baseline (EBA), attributed to the fully parallel OT/Sinkhorn computation on GPUs.
💻Code: github.com/ZW471/PLASMA-Prot…
📜Paper: arxiv.org/abs/2510.11752
#ProteinStructure #StructuralBioinformatics #OptimalTransport #DeepLearning #ICLR2026 #ProteinMotifs #FunctionalAnnotation #ComputationalBiology
3
25
2,033
Tokenizing Loops of Antibodies
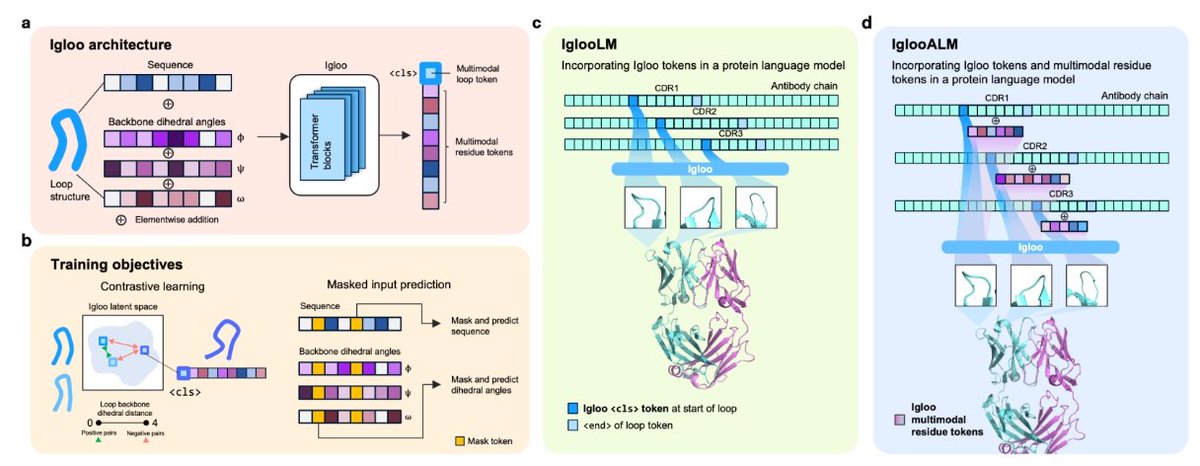
1. The paper introduces Igloo (ImmunoGlobulin LOOp Tokenizer), a multimodal tokenizer that represents antibody CDR loops at the loop/substructure level (not residue level), encoding both sequence and backbone dihedral angles (phi/psi/omega) into a loop token.
2. Core idea: train a self-supervised transformer with a contrastive objective so that loops with similar backbone dihedral-angle patterns map close in latent space. Similarity is defined using a dihedral distance metric (preferred over RMSD because RMSD can miss 180° dihedral flips that invert side-chain orientations).
3. Igloo addresses a long-standing limitation of canonical CDR clustering: incomplete coverage (e.g., many H3 loops do not map to canonical clusters). Igloo assigns tokens to all loops while still recovering canonical conformations for 90.6% of SAbDab loops that have canonical labels.
4. Training scale and data mix: 807,815 loop regions total, combining 108,167 experimentally resolved loops (SAbDab STCRDab) and 699,648 predicted loops from paired OAS sequences via Ibex structure prediction, with careful sequence-identity splits to reduce leakage.
5. Architecture and objectives: dihedral angles are embedded via unit-circle features (cos/sin), added to amino-acid embeddings, then processed with a BERT-style transformer using a learnable <cls> token as the loop representation. Training uses (a) multimodal masking and reconstruction, (b) dihedral-distance contrastive learning with positive/negative pairs, and (c) a vector-quantized codebook to produce discrete tokens for fast lookup.
6. Structure retrieval benchmark (paratope retrieval): given a query loop, retrieve the closest loops from a structural database. Igloo achieves state-of-the-art Precision@20 across CDRs, and for the hardest loop (H3) improves retrieval of similar dihedral backbones by 6.1% over the prior best baseline (0.402 vs 0.379 for D < 0.47).
7. Discrete-token clustering: without being given loop-type annotations, the learned codebook clusters are highly homogeneous by loop type and length (type purity 0.983; length purity 0.965), and show strong agreement with established canonical cluster partitions.
8. Igloo tokens as special tokens in antibody language models: IglooLM inserts a loop token at each CDR boundary and fine-tunes from IgBert (420M). On AbBiBench binding-affinity prediction for heavy-chain variant sets, IglooLM beats the base IgBert model on 8/10 antibody-antigen targets and performs comparably to much larger models (including ones with ~7x more parameters).
9. Generative design with structural control: IglooALM combines loop tokens plus residue-level multimodal tokens to sample loop sequences that are diverse yet structurally consistent. Compared to antibody inverse-folding baselines (AbMPNN, AntiFold), IglooALM generates loops with better self-consistency RMSD across many sequence-identity bins; a SARS-CoV-2 H3 redesign example reports ~0.27 sequence identity while staying <1 Å RMSD to the original loop.
10. Library prioritization at scale (zero-shot): on a HER2 CDR H3 mutagenesis library (38,860 variants), selecting sequences that share the same discrete Igloo token as a seed yields 55.3% binders, a ~1.9x enrichment over the library baseline (29.1%), and runs quickly (minutes) using sequence-based tokenization.
💻Code: github.com/prescient-design/…
📜Paper: tandfonline.com/doi/full/10.…
#AntibodyEngineering #ProteinLanguageModels #CDR #StructuralBioinformatics #ContrastiveLearning #GenerativeAI #ComputationalBiology #Therapeutics
2
23
1,535
Tokenizing Loops of Antibodies
1. The paper introduces Igloo (ImmunoGlobulin LOOp Tokenizer), a multimodal tokenizer that represents antibody CDR loops at the loop/substructure level (not residue level), encoding both sequence and backbone dihedral angles (phi/psi/omega) into a loop token.
2. Core idea: train a self-supervised transformer with a contrastive objective so that loops with similar backbone dihedral-angle patterns map close in latent space. Similarity is defined using a dihedral distance metric (preferred over RMSD because RMSD can miss 180° dihedral flips that invert side-chain orientations).
3. Igloo addresses a long-standing limitation of canonical CDR clustering: incomplete coverage (e.g., many H3 loops do not map to canonical clusters). Igloo assigns tokens to all loops while still recovering canonical conformations for 90.6% of SAbDab loops that have canonical labels.
4. Training scale and data mix: 807,815 loop regions total, combining 108,167 experimentally resolved loops (SAbDab STCRDab) and 699,648 predicted loops from paired OAS sequences via Ibex structure prediction, with careful sequence-identity splits to reduce leakage.
5. Architecture and objectives: dihedral angles are embedded via unit-circle features (cos/sin), added to amino-acid embeddings, then processed with a BERT-style transformer using a learnable <cls> token as the loop representation. Training uses (a) multimodal masking and reconstruction, (b) dihedral-distance contrastive learning with positive/negative pairs, and (c) a vector-quantized codebook to produce discrete tokens for fast lookup.
6. Structure retrieval benchmark (paratope retrieval): given a query loop, retrieve the closest loops from a structural database. Igloo achieves state-of-the-art Precision@20 across CDRs, and for the hardest loop (H3) improves retrieval of similar dihedral backbones by 6.1% over the prior best baseline (0.402 vs 0.379 for D < 0.47).
7. Discrete-token clustering: without being given loop-type annotations, the learned codebook clusters are highly homogeneous by loop type and length (type purity 0.983; length purity 0.965), and show strong agreement with established canonical cluster partitions.
8. Igloo tokens as special tokens in antibody language models: IglooLM inserts a loop token at each CDR boundary and fine-tunes from IgBert (420M). On AbBiBench binding-affinity prediction for heavy-chain variant sets, IglooLM beats the base IgBert model on 8/10 antibody-antigen targets and performs comparably to much larger models (including ones with ~7x more parameters).
9. Generative design with structural control: IglooALM combines loop tokens plus residue-level multimodal tokens to sample loop sequences that are diverse yet structurally consistent. Compared to antibody inverse-folding baselines (AbMPNN, AntiFold), IglooALM generates loops with better self-consistency RMSD across many sequence-identity bins; a SARS-CoV-2 H3 redesign example reports ~0.27 sequence identity while staying <1 Å RMSD to the original loop.
10. Library prioritization at scale (zero-shot): on a HER2 CDR H3 mutagenesis library (38,860 variants), selecting sequences that share the same discrete Igloo token as a seed yields 55.3% binders, a ~1.9x enrichment over the library baseline (29.1%), and runs quickly (minutes) using sequence-based tokenization.
💻Code: github.com/prescient-design/…
📜Paper: tandfonline.com/doi/full/10.…
#AntibodyEngineering #ProteinLanguageModels #CDR #StructuralBioinformatics #ContrastiveLearning #GenerativeAI #ComputationalBiology #Therapeutics
3
9
940