
EMReady2: Improvement of cryo-EM and cryo-ET maps by local quality-aware deep learning with Mamba
1. EMReady2 is a real-space, single-map post-processing method that targets a key pain point in cryo-EM/cryo-ET interpretation: strong local heterogeneity (variable local resolution, anisotropy, fragmented connectivity) that makes a “one-size-fits-all” sharpening strategy unreliable.
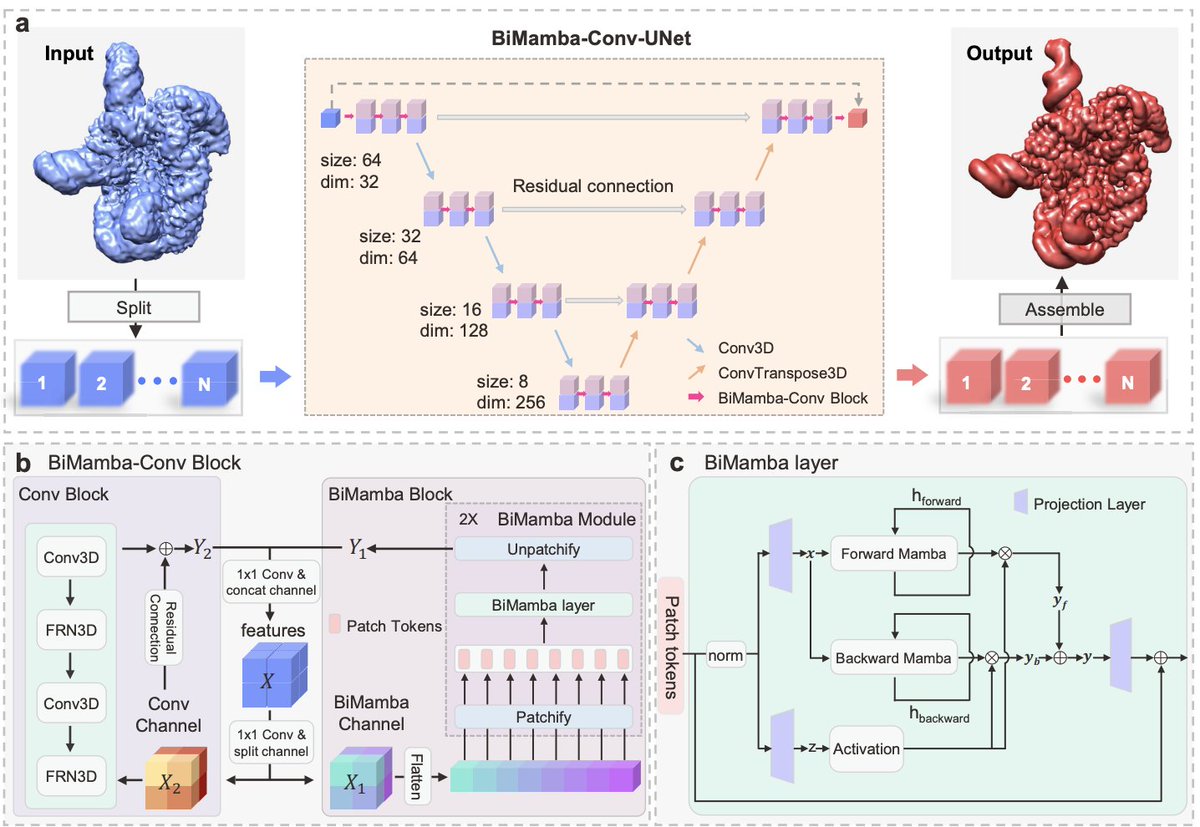
2. The main architectural shift is BiMCUnet, a dual-branch UNet that fuses a bidirectional Mamba (state-space) branch for long-range/global context with a convolutional branch for local detail, aiming to keep global coherence while restoring local connectivity—without the quadratic cost of attention.
3. A core training innovation is local quality-aware supervision: instead of generating targets with a uniform resolution, EMReady2 builds simulated maps using atom-wise local resolution estimated from Q-scores, then blends local and global resolution (w=0.8). This is designed to reduce bias from poorly modeled regions and better match real map heterogeneity.
4. The training set is substantially broadened versus the original EMReady: balanced inclusion of proteins and nucleic acids, plus medium/low-resolution cases (6–10 Å) and cryo-ET subtomogram averaging (STA) maps—expanding the practical scope to 2–10 Å and to RNA/DNA-heavy assemblies.
5. On 118 diverse cryo-EM SPA maps (2–10 Å), EMReady2 improves map–model FSC-0.5 for 113/118 cases and leads across multiple metrics (mean FSC-0.5 4.65 Å vs 5.78 Å deposited; Q-score 0.493 vs 0.454; main-chain Q-score 0.557 vs 0.491), outperforming DeepEMhancer, phenix.auto_sharpen, EMReady, and CryoTEN in the reported benchmarks.
6. For nucleic-acid–rich maps (>10% nucleic acids; 18 cases), EMReady2 shows larger gains, consistent with the expanded training distribution (mean FSC-0.5 4.20 Å vs 5.70 Å deposited; Q-score 0.499 vs 0.453; main-chain Q-score 0.543 vs 0.462). Segmented evaluation suggests improvements are even stronger within nucleic-acid regions than globally.
7. EMReady2 extends to cryo-ET STA: on 18 STA maps (3–10 Å), it improves masked FSC-0.5, Q-score, CC_mask, and main-chain Q-score versus deposited maps and competing post-processing tools, supporting use beyond SPA where maps are typically lower resolution and noisier.
8. Interpretability is evaluated via automated de novo model building (phenix.map_to_model) for maps better than 5 Å: across 832 chains, models built from EMReady2-processed maps show higher residue coverage and sequence recall for both proteins (coverage 70.51%, recall 32.24%) and nucleic acids (coverage 72.14%, recall 27.40%) than deposited maps and other post-processing methods.
9. The paper explicitly probes “hallucination” risk on intrinsically disordered regions and reports that EMReady2 improves contrast/connectivity without forcing ambiguous density into canonical helices/sheets; it also emphasizes that conformational/compositional heterogeneity must be handled upstream, and that small ligands/ions/waters remain challenging for data-driven enhancement.
10. Efficiency is a practical highlight: average runtime across 136 maps is reported as 14.81 s, faster than CryoTEN (17.33 s), and far faster than EMReady (83.26 s) and DeepEMhancer (90.98 s), aligning with the linear-scaling motivation for adopting Mamba-style state-space modeling.
💻Code: github.com/huang-laboratory/…
📜Paper: doi.org/10.1038/s41467-026-7…
#cryoEM #cryoET #structuralbiology #computationalbiology #deeplearning #Mamba #statespacemodels #UNet #mapsharpening #nucleicacids
4
31
2,759