
No longer active here. Find me on Bluesky: bsky.app/profile/rrwick.bsky…
Joined August 2013
- Tweets 978
- Following 383
- Followers 3,141
- Likes 1,506
64 Photos and videos





This will be my last post here. I'm moving to Bluesky:
bsky.app/profile/rrwick.bsky…
Hope to see you there!
4
427
Do you make core genome alignments for phylogenomics? @monaltaouk and I explored how including sites with some missing data (a soft core) can improve analysis, especially for large datasets.
microbiologyresearch.org/con…
(1/4)
2
30
90
5,774
To make producing soft core alignments easier, we developed Core-SNP-filter, a simple and efficient tool to process SNP alignments with user-defined thresholds.
github.com/rrwick/Core-SNP-f…
(4/4)
8
475
New year, new assemblies!
I'm excited to announce Autocycler, my new tool for consensus assembly of long-read bacterial genomes!
It's the successor to Trycycler, designed to be faster and less reliant on user intervention.
Check it out: github.com/rrwick/Autocycler…
(1/5)
5
117
261
15,680
Just had my first experience with CycloneSEQ data and shared my findings in this blog post:
rrwick.github.io/2024/12/17/…
How does CycloneSEQ compare to @nanopore? I looked at both read-level and consensus-level accuracy. Check it out!
1
9
35
1,891
Ryan Wick retweeted

18 Oct 2024
If you want to try out the new --bacteria Medaka polishing option after reading Ryan's thoughts in his latest blogpost, the newest Hybacter v0.10.0 release now includes it as the default
github.com/gbouras13/hybract…

A little bit overdue, but here are my thoughts on @nanopore's recent release of Medaka:
rrwick.github.io/2024/10/17/…
15
50
5,156
A little bit overdue, but here are my thoughts on @nanopore's recent release of Medaka:
rrwick.github.io/2024/10/17/…
1
31
105
12,124
Ryan Wick retweeted

12 Sep 2024
Excited to share this preprint where we’ve used genomic analysis of >3000 genomes to study Klebsiella pneumoniae in a One Health perspective 🦠🔬👩💻#OneHealth #KLEBGAP #KlebClub @IrenLohr @DrKatHolt @NORKLEBNET
12 Sep 2024

A genome-wide One Health study of Klebsiella pneumoniae in Norway reveals overlapping populations but few recent transmission events ... biorxiv.org/cgi/content/shor… #biorxiv_micrbio
2
23
67
6,802
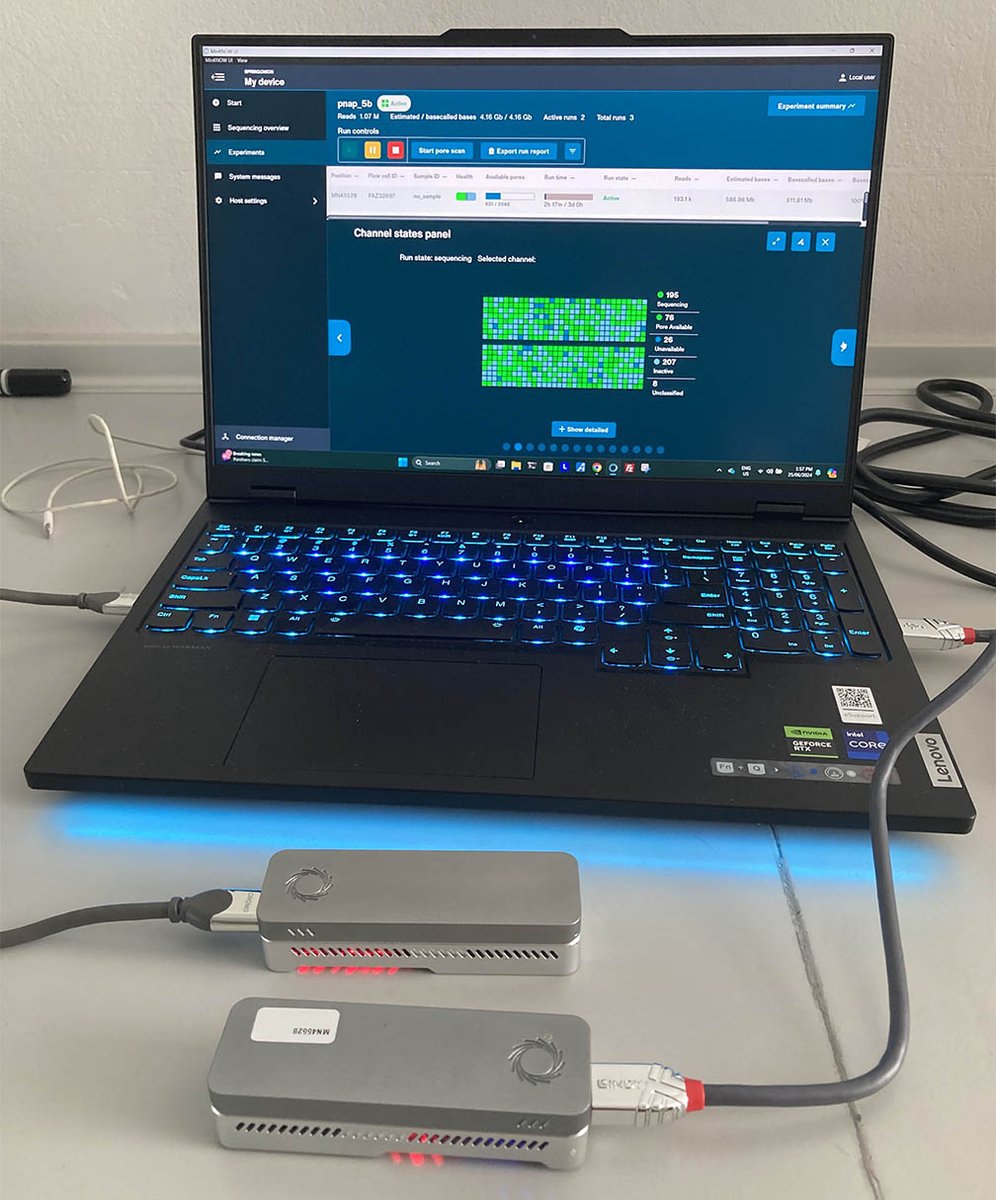
Our OnION sequencing computer now has a little sibling: Spring OnION!
Short post with some stats and benchmarks: rrwick.github.io/2024/08/16/…
4
8
93
4,940
I finally got around to testing HERRO (github.com/lbcb-sci/herro), the new @nanopore read correction algorithm which takes simplex ONT reads to PacBio-HiFi-level accuracy.
I was impressed! Findings are on my blog in a two-part post:
rrwick.github.io/2024/07/26/…
rrwick.github.io/2024/07/31/…
3
75
226
20,420
Ryan Wick retweeted

22 Jul 2024
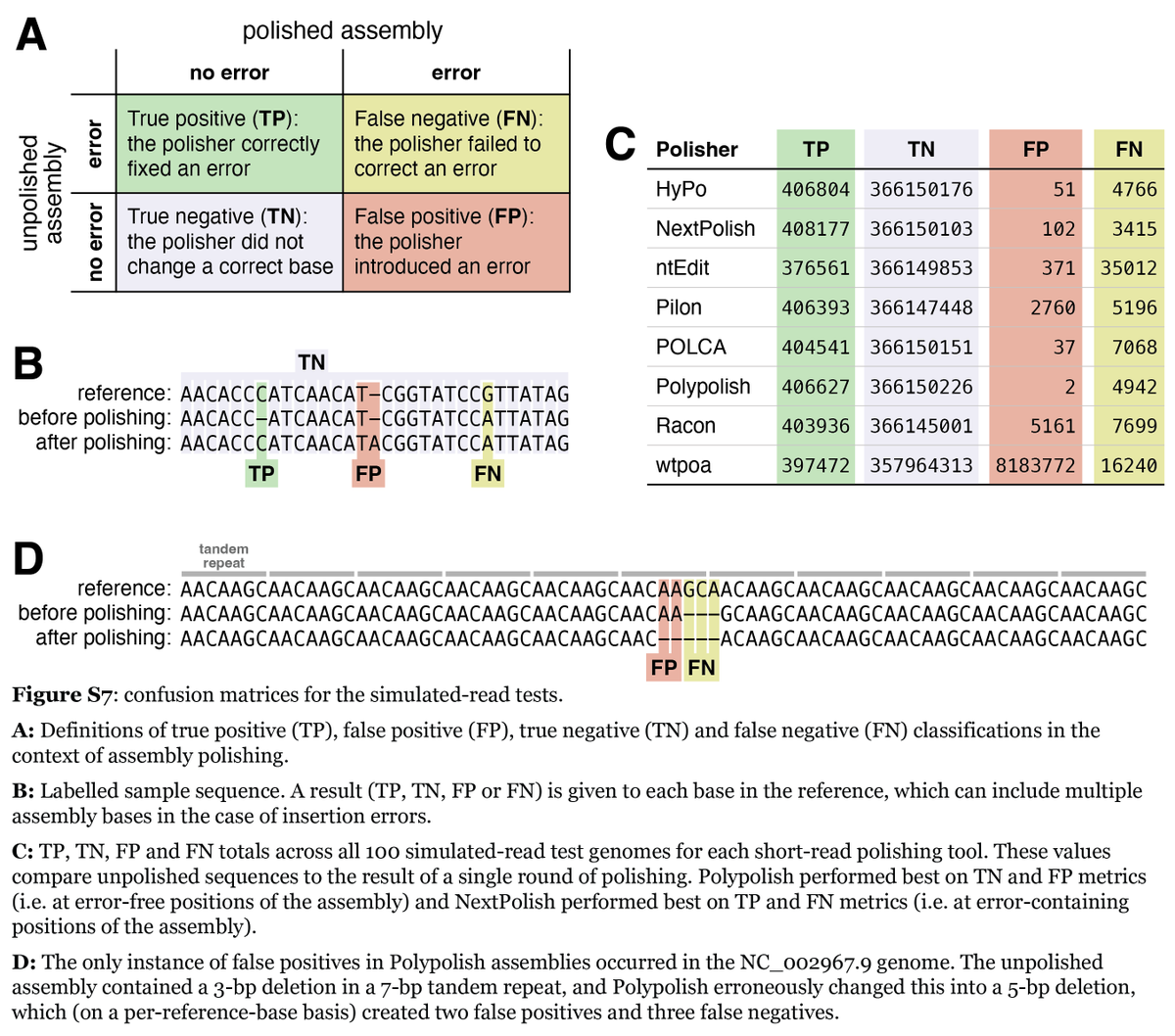
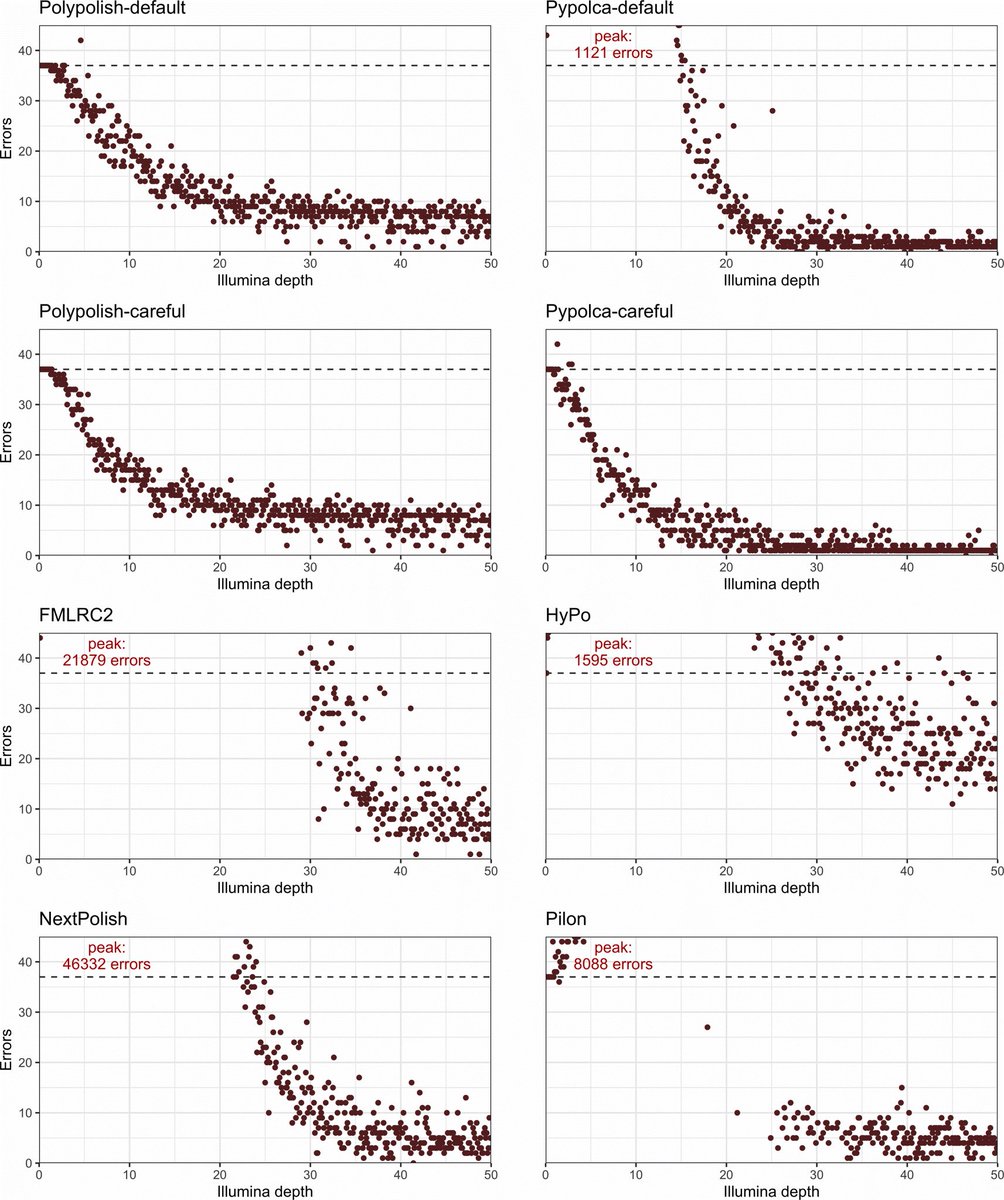
How low can you go? Short-read polishing of Oxford Nanopore bacterial genome assemblies. Published #OpenAccess and fee-free in #MGen using a #PublishAndRead agreement: doi.org/10.1099/mgen.0.00125…
9
24
4,532
Ryan Wick retweeted
15 Jul 2024
It has been public for a while, but with #VoM2024 starting, today is as good as any to introduce our new tool phage genome annotation tool Phold. Phold uses structural homology to assign functions to phage proteins (1/n)
1
62
211
26,145
Ryan Wick retweeted

28 Jun 2024
I am excited to win a CZI EOSS grant! This is the first grant I have ever won after 20 years in science. It feels wonderful to finally be funded for my open-source contributions. With @wytamma we will deliver the next generation of my popular Snippy software! 🦘🦘🦘

Open source tools are crucial for biomedical research. We're collaborating with @KavliFoundation @Wellcometrust to support developers & maintainers of critical #OpenSource projects, promoting innovation & community engagement in the field. More ▶️ czi.co/3zjN56l

40
49
354
23,372
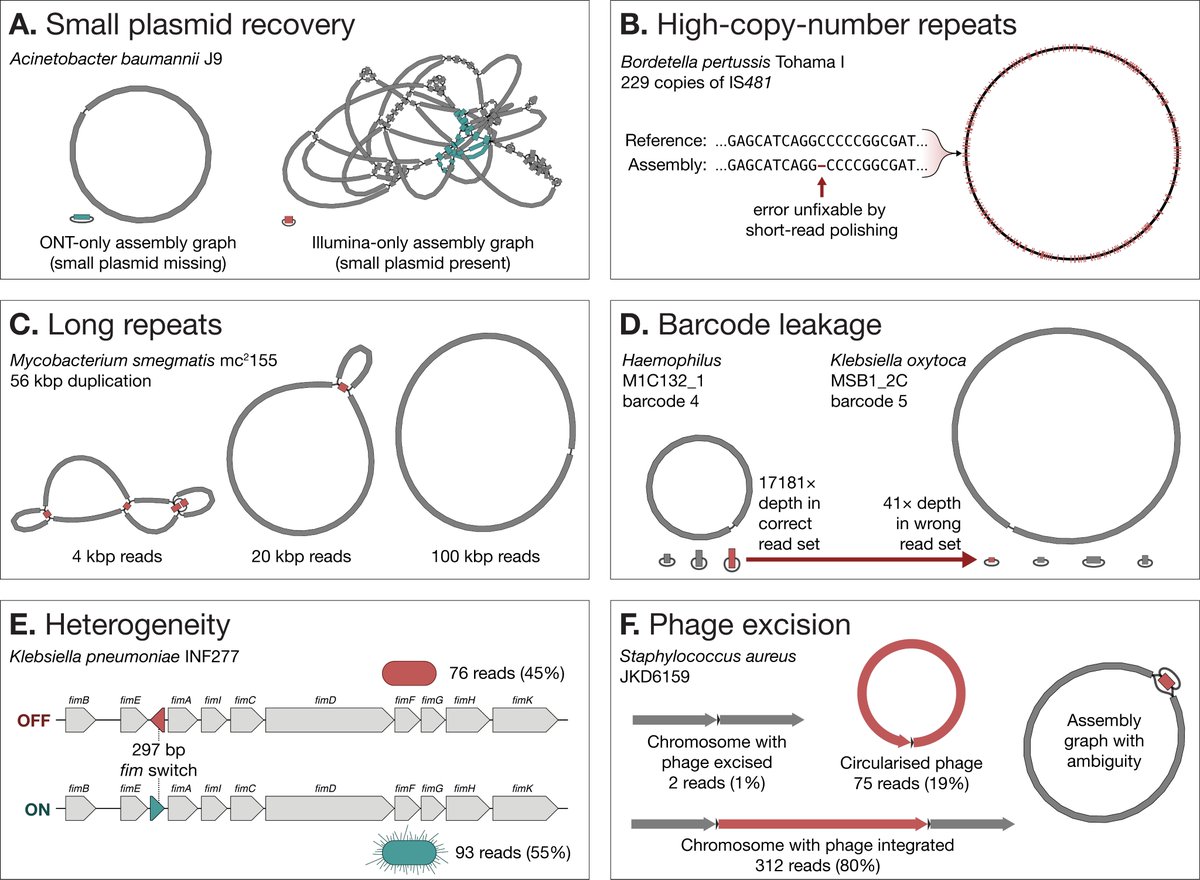
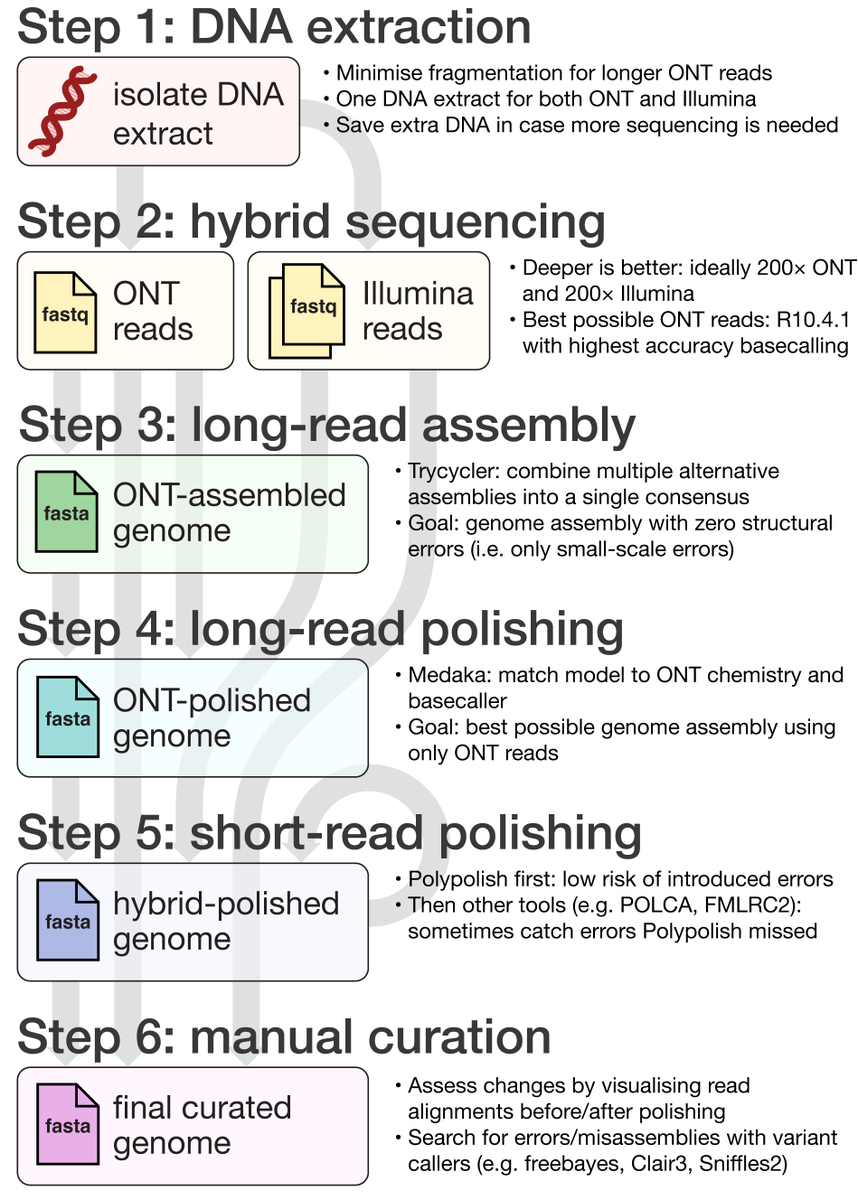
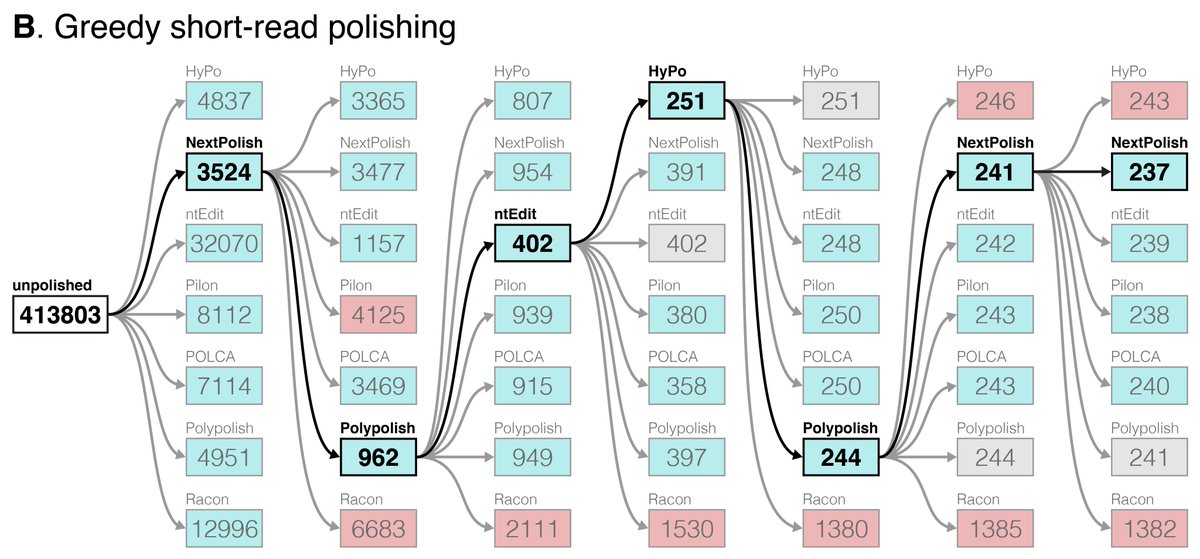
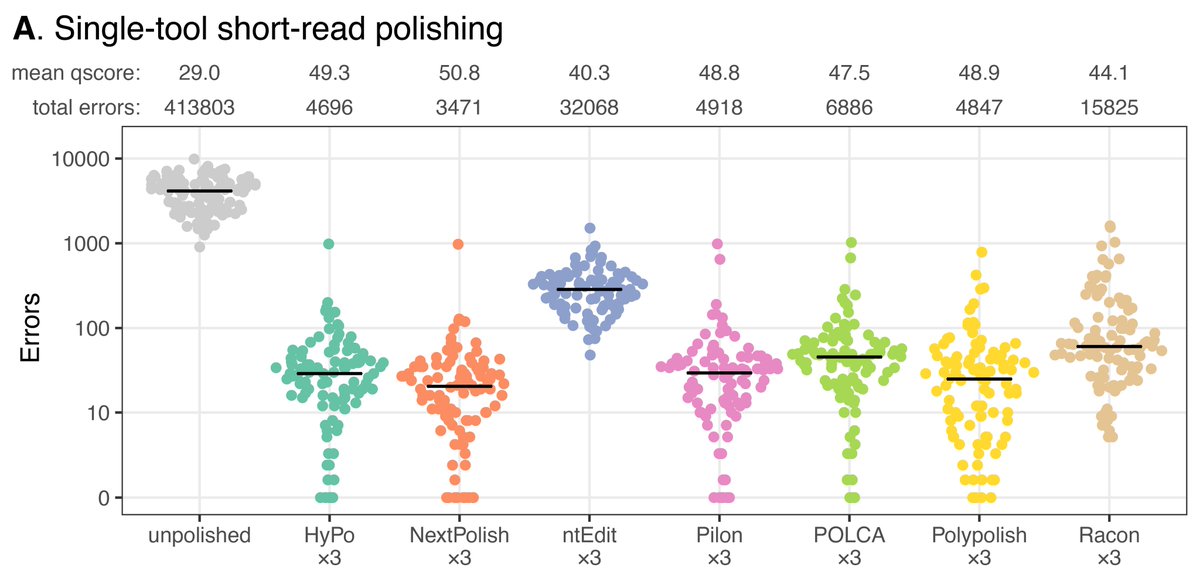
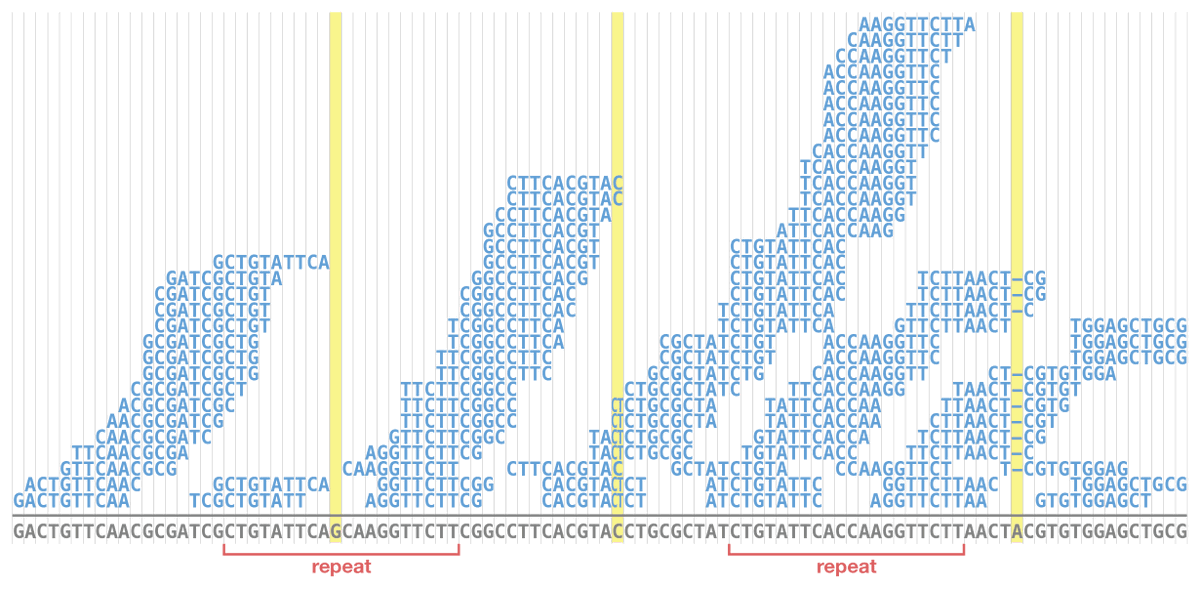
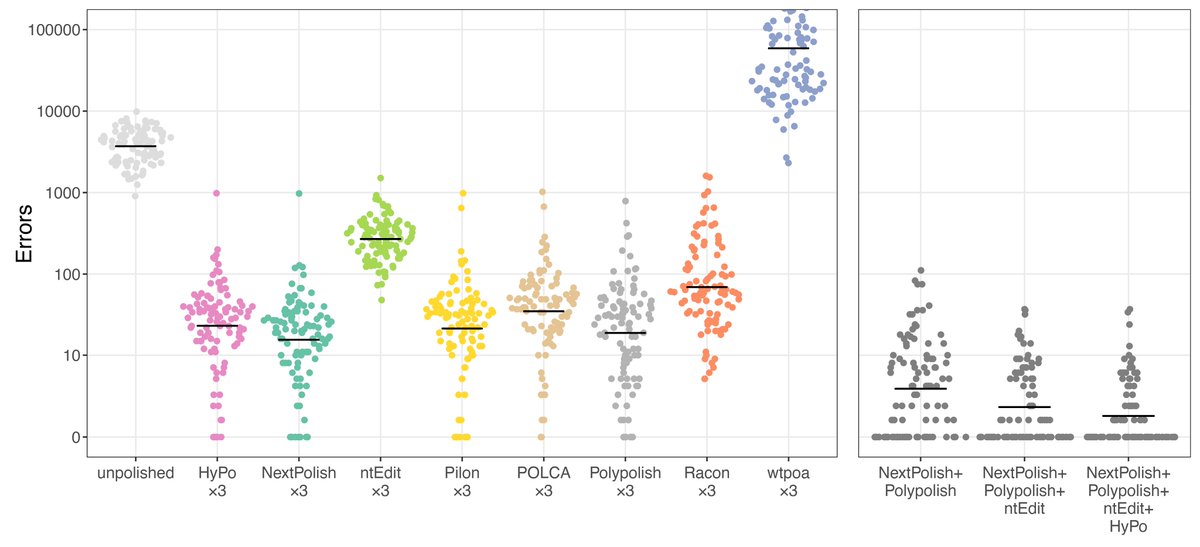
New blog post:
rrwick.github.io/2024/06/11/…
ONT-only bacterial assemblies are much better than they were only a couple of years ago. Often <10 errors in the whole genome. Does that mean that less short-read sequencing is required for polishing? See my post for the answer!
(1/4)
2
55
153
17,261
The paper also introduces George's tool Pypolca:
github.com/gbouras13/pypolca
This is a reimplementation of the POLCA polisher (by @AlekseyZimin5) that is easier to install and run. Pypolca also has some new options (e.g. --careful) which give it more flexibility than POLCA.
(3/4)
1
12
1,017