
Joined March 2021
- Tweets 1,555
- Following 1,135
- Followers 546
- Likes 82,725
56 Photos and videos












Pinned Tweet

21 May 2024
The first paper of my graduate school career has been published!! Drake Phelps and I, along with our co-authors Giuliano Ferrero, Jamie Dewitt, and Jeff Yoder review how #PFAS impact innate #immune function across species. Please give it a read!
doi.org/10.1080/1547691X.202…
2
4
20
2,192
Ashley Connors retweeted

May 17
People can spread this version of hantavirus to each other. What makes Andes virus special?
sciencenews.org/article/ande…
15
113
379
114,508
Ashley Connors retweeted

Attention @arxiv authors: Our Code of Conduct states that by signing your name as an author of a paper, each author takes full responsibility for all its contents, irrespective of how the contents were generated. 1/
139
920
6,557
1,089,157
Ashley Connors retweeted

The recent U.S. FDA characterization of baby formula as “safe” is entirely misleading.
In the FDA dataset, about two-thirds of the infant formulas tested had detectable PFAS. The highest levels were found in soy-based formula samples, especially powder soy-based formulas.
These detections were mainly driven by PFBA, with some soy powder samples containing around 30-34 ppt.
@bennettpeer @Nathan_Donley
theguardian.com/us-news/2026…
5
6
203
Ashley Connors retweeted

May 7
FYI - People are confusing "hantavirus has a long incubation period" with "covid was contagious before symptoms showed up."
Incubating ≠ contagious.
Andes virus is not an upper-airway, high-shedding respiratory virus. It evolved for a very different survival strategy.
39
269
7,225
397,105
Ashley Connors retweeted

I think we can all agree the government telling scientists what to publish is catastrophically bad. When I say they're annihilating American science, this is what I mean.
May 5

Breaking News: The FDA has blocked publication of research that found widely used Covid-19 and shingles vaccines were safe. nyti.ms/49dtF24
16
141
726
16,398
Ashley Connors retweeted

15 Dec 2025
Hi, medical toxicologist/ER doctor here 👋
here’s what you need to know about medetomidine in the illicit opioid supply

9
46
344
32,486
Ashley Connors retweeted

Apr 18
The hill I will die on - we have to rethink graduate training.
“Scientists are trained for a world where data speaks for itself. Where misinformation moves slowly. Where scientific expertise naturally rises above noise. That world is gone.”
sciencepolitics.org/2026/03/…

55
231
836
104,643
Ashley Connors retweeted

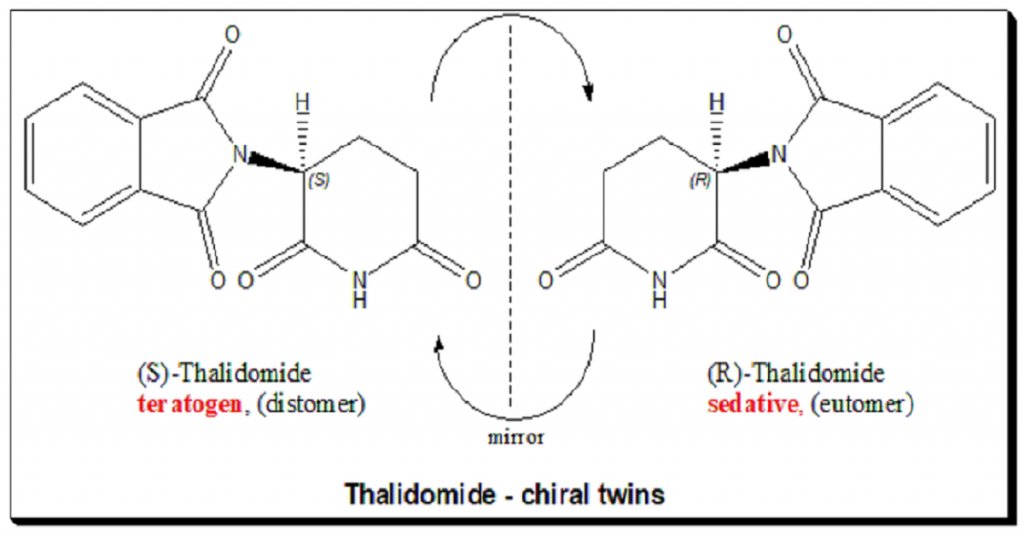
In the late 1950s, a drug called thalidomide was widely prescribed in Europe and Australia as a highly effective treatment for morning sickness in pregnant women. The manufacturer assured doctors that the drug was completely safe.
However, thalidomide is a chiral molecule, meaning it exists in two mirror-image forms. While one form successfully cured nausea, the other was highly teratogenic. The drug crossed the placental barrier and interfered with the development of fetal blood vessels, leading to a global catastrophe. Over 10,000 babies around the world were born with severe limb deformities, often missing their arms and legs entirely.
The United States largely avoided this tragedy thanks to an FDA inspector named Frances Kelsey, who famously stonewalled the drug's approval by demanding more safety data, despite facing massive pressure from the manufacturer.
This averted disaster directly resulted in the passage of the 1962 Kefauver-Harris Amendment, which fundamentally defined the modern regulatory framework for the FDA. Prior to this amendment, the regulatory bar was essentially just proving that a drug wouldn't acutely kill people (kind of like the current bar for most recreational peptides). The 1962 amendment introduced a much higher standard: pharmaceutical companies were now legally required to conduct controlled clinical trials to prove that their drugs were not only safe, but also actively effective for their intended use.
The gap between the old standard and this new requirement was massive. In fact, roughly two-thirds of the drugs that had previously passed the 1938 safety requirements ended up failing the new 1962 efficacy bar. This historical context is often cited by public health experts today when defending the FDA's grueling, multi-phase clinical trial requirements against critics who argue for faster drug approvals or unregulated access to experimental compounds.
3
8
52
4,202
Ashley Connors retweeted

Apr 8
Excited to share our new paper! 🐝🎉Chronic exposure to the insecticide sulfoxaflor alters gene expression in bumblebee ovaries, suppresses reproduction, and changes behavior—ultimately impairing colony function.
Take a look:
sciencedirect.com/science/ar…
1
2
42
Ashley Connors retweeted

Apr 1
Columbia and NYU researchers tracked 11,000 people for 20 years.
Urine tests before. Urine tests after.
When they switched to clean water, their risk of dying from cancer and heart disease dropped 50%.
Here's what was in their water the entire time: 🧵


3
53
234
23,814
Mar 15
A good reminder that regulations are written in blood.

wdym they hired young women in their 20s to work with radium, told them it was harmless, all while the men handling the same material used protective gears, and when the women’s bodies started breaking down ( jaws falling, gums splitting, bones fracturing, organs failing), the company claimed the women actually had syphilis from immorality ???
27
Mar 11
RT @Feisty_Waters: No AI, I don’t want you to summarize everything. I want to read things and think about them with my own brain.
1,063
Ashley Connors retweeted

Mar 6
100% correct.
Mar 5

Alcohol is a million times more dangerous to your health than seed oils, sugar, or high fructose corn syrup
If you're hating on any of those 3 things and you drink alcohol regularly, you're focusing on the WRONG things
2
2
6
358
Ashley Connors retweeted

Doctors and parents should be aware that the medical textbook description of a "bright red measles rash" is based on white skin. On brown or black skin, the measles rash can be much harder to see. It may appear as darker, dusky or purple patches rather than bright red spots.
32
1,594
4,431
60,229
Ashley Connors retweeted

Feb 13
A common misconception I am seeing in the replies is that antibiotic resistance isn’t about your body becoming resistant.
It’s the bacteria that become resistant.
Even if you personally use antibiotics responsibly, or have never used antibiotics ever , if those resistant bacteria infect you, the standard antibiotics won't work on you too.
That is why it is a community problem and needs urgent awareness programs for both patients and healthcare professionals
Feb 13

Antibiotic resistance means one day
a simple cut could kill like it did 100 years ago.
37
807
3,991
158,019
Ashley Connors retweeted

“No one expected this.”
Jane Hoppin and fellow scientists were surprised to find TFA in years-old samples, raising new questions about PFAS and environmental health.
Learn more. ➡️: ncst.at/hYy950Y7JK4

1
1
84
19
Use a broom or your kid if you have to, but clear it off!

PSA to all Charlotte drivers who are not used to the snow: clean off the tops of your cars before you drive or else it will fly back and hit the drivers behind you‼️‼️‼️
30
Learning to evade the immune system is one of the key developments of cancer cells.
Before that point, cancer cells can be destroyed by the immune system because they stop expressing the “self” signals that immune cells use to identify which cells are on the home team.

Among the things this misses: cancer is bad because it's *your own cells*, so your immune system doesn't see it as a threat.
Even if cultured meat cells grew well (they don't 😭), anyone too immunocompromised to reject *cow cells* would have died long ago of bacterial infection
1
53
In relation to lab grown meat, also consider that you’d be chewing it up and dropping it into a vat of acid. Most cells wouldn’t survive that. The ones that do are usually bacteria, because many are adapted to extreme environments.
1
15