
Tech entrepreneur empowering people to live longer, freer, happier lives - CoFounder of Humanity Inc crunchbase.com/person/mike-g…
Joined August 2008
- Tweets 3,226
- Following 2,637
- Followers 3,340
- Likes 1,420
50 Photos and videos














Michael Geer retweeted

7 Jul 2025
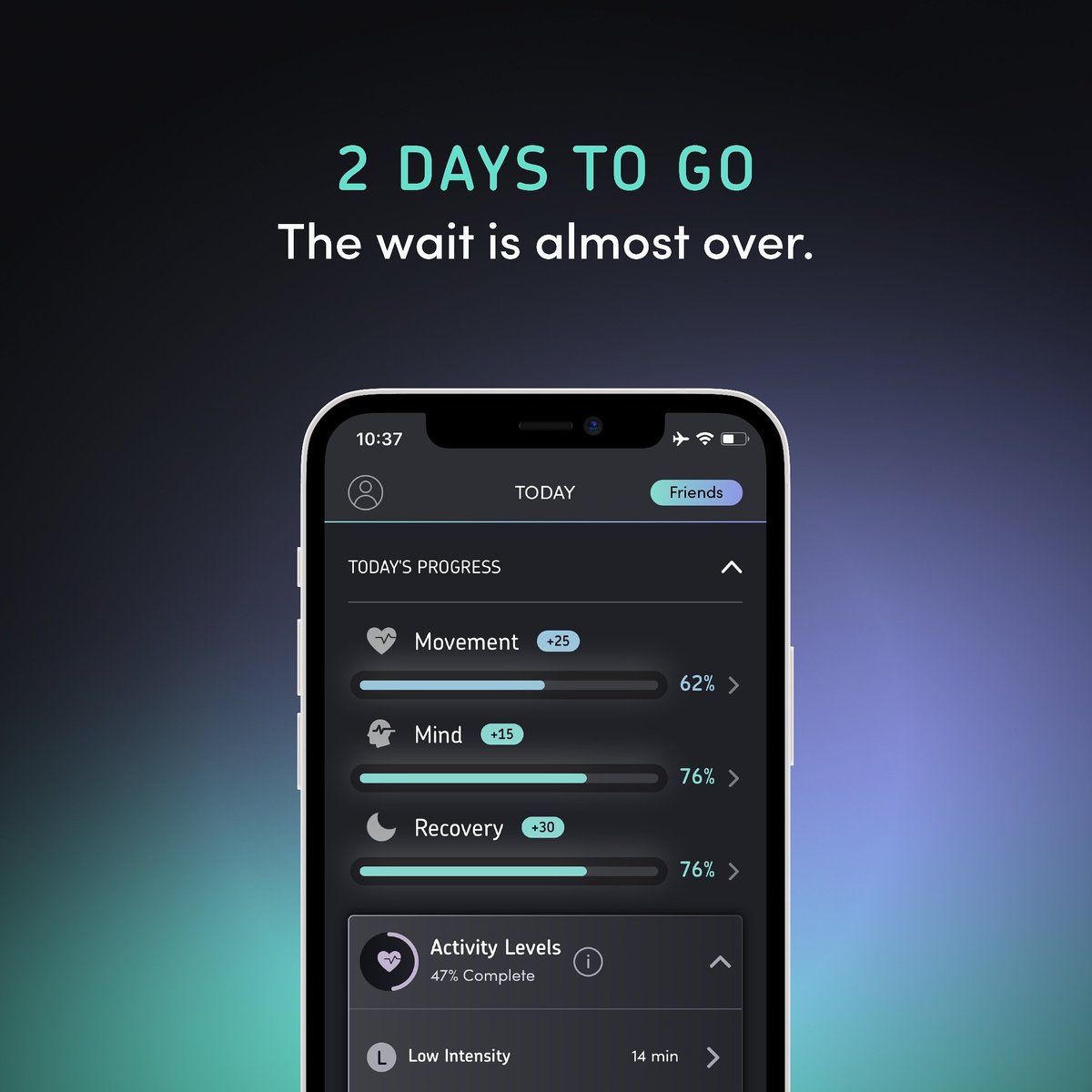
Level 1: Tackling individual hallmarks of aging, like metabolic health or inflammation (adding years to life).
Level 2: Stabilizing biological systems against random fluctuations—reducing physiological “noise” that destabilizes health as we age (adding decades of healthy life).
Level 3: The ultimate frontier: stopping aging altogether, opening the possibility of radically extending lifespan.
Our current collaboration with Chugai aims to excel at Level 1. This allows us to focus in-house on developing a proof-of-concept Level 2 intervention. The road to Level 3 remains sketchy and certainly longer, but it’s no longer just a dream.
And when we launched Gero, it was with the quiet hope that, maybe, if we work hard enough, fast enough, and wisely enough, the question from a child might one day have a better answer.
Today, we’re one step closer to building a no-nonsense intervention against aging in our species.
Many people ask me: “What can I do to help stop aging?”
Now you have a simple answer—and it matters.
If you're an investor who sees the opportunity in longevity—please get in touch!
If you're a scientist, engineer, or drug developer eager to work on the frontiers of aging biology—join us!
And if you believe aging should be treated—share this post, comment, or tag someone who should see it -- anyone who may share this vision.
globenewswire.com/news-relea…
endpoints.news/roche-backed-…
fiercebiotech.com/biotech/ro…
lifespan.io/news/playing-the…
#Longevity #Aging #AI #DrugDiscovery #FutureOfMedicine #ChasingDreams #Entropy
2/2
7
22
119
4,763
Michael Geer retweeted
7 Jul 2025
BREAKING: Gero Signs Potential Multi-Billion-$ Deal to Fuel Our Mission to Stop Aging --
A few years ago, my son, aged five at the time, asked what happened to the dinosaurs. After learning their sad story, he told me he wanted to fly to the Moon and defend Earth from falling comets. He began assembling his crew—quickly running out of family members and turning to his grandparents—but then stopped and asked: “Dad... Grandpa and Grandma are already old. It will be hard for them to shoot down comets. Can we make it so people don’t age?”
At that tender age, he couldn’t yet fully grasp death, but he already understood aging—and its cruel way of stealing our strength and time. This story has played out billions of times in human history: a child realizes, for the first time, that life is finite. But maybe—just maybe—his generation will be the first to hear a different answer.
Today, we took a step toward that answer. Together with Chugai Pharmaceutical (a member of Roche group), we announced a partnership to develop therapies that target the root causes of major age-related diseases. At Gero, we’ve spent the last 15 years building physics-informed AI models, trained on massive longitudinal datasets tracking human biology over years. These models revealed something critical: in long-lived mammals like humans, aging is mostly an entropic process—slow, irreversible damage that accumulates over time. But diseases? They are failures of individual systems—failures that occur faster in aging bodies but, crucially, are still actionable. Some of these failure points are shared across multiple diseases, and targeting these shared vulnerabilities could lead to therapies that fight multiple diseases at once.
When we started our journey, there were no examples of success to follow. What we had was the belief that science works—and that complex phenomena like aging and chronic diseases could be understood through first principles borrowed from other sciences like physics, and with the help of emerging AI and machine learning tools. We also had no clear idea how to fund such an enterprise. Our inspiration came from nature itself—from mammals like naked mole rats and bats, which show little to no signs of aging over long lifespans.
Things are moving fast. The recent success of GLP-1 drugs (Ozempic, Wegovy) has already shown what’s possible: a single drug class improving metabolic health, heart disease, obesity, and more. What once seemed like science fiction is now reality. And yet, as I argued in my Lifespan's column, these are still just our first flying machines. The real challenge is to build the rockets—to address the fundamental physics of aging, not just its symptoms.
Our models predict three levels of intervention against aging in humans (as always, please do not forget to like, follow and repost before scrolling down for more explanations and links):
1/2

54
89
513
83,949
Michael Geer retweeted
23 Jun 2025
Designing small molecules to hit protein targets is the holy grail of drug discovery, but it’s a beast of a problem. For years, I’ve been obsessed with finding a practical way to crack it. Coming from a physics background, my gut always leaned toward modeling—think docking molecular dynamics (MD) with explicit or implicit water. Sounds cool, right? Problem is, these methods often fall flat.
Why? First, there’s the energy scale mess: two charges at 1Å interact with a whopping 15eV (170,000K), while a good ligand’s binding free energy is 50kJ/mol (600K). That’s a massive gap. Water’s dielectric susceptibility (100x attenuation) tries to bridge it, but that’s a bulk property—useless in a tiny binding pocket. Proteins are floppy, teetering just a few degrees from denaturation, constantly shape-shifting in ways Nature exploits to control molecular activity, so forget assuming they have a small dielectric constant. But, like with water, this doesn’t help much in a small pocket either.
More headaches? Atomic charges aren’t fixed. Polarization effects aren't small and require full quantum calc to get right. Even such "tiny" errors in a decent-sized ligand snowball fast, making the problem a computational nightmare. Sure, you can dock and maybe find something, but often it’s easier to just screen compounds and call it a day.
But what if we’re asking the wrong question? Nature faced this same puzzle—evolution had to optimize molecular machinery to churn out biologically active molecules. All the “useful” chemistry might already be encoded in our genome. What if predicting arbitrary ligand-protein interactions is a fool’s errand? Instead, Nature might have cherry-picked proteins that reliably bind certain molecule classes, with smooth “medchem” tweaks to fine-tune interactions over time.
If that’s true, the rules for crafting bioactive molecules are written in our genome—a kind of chemical language. Enter modern generative AI: what if we could “listen” to the stories in biological sequence data (genome, proteome) and learn to speak chemistry, spitting out molecules that play nice with proteins? If the physics part is solved by the Natural selection in some practical way, would we expect that finding biologically active molecules is not a physics but rather is a language problem?
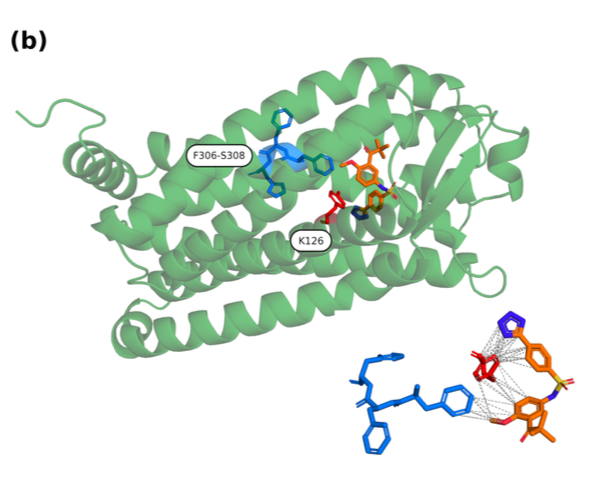
That’s the spark behind our new model, ProtoBind-Diff, a structure-free masked diffusion model that generates molecules conditioned directly on protein sequences via pre-trained language model embeddings. Trained on over a million protein-ligand pairs from BindingDB, it pumps out chemically valid, novel, and target-specific ligands without ever needing 3D structural data. In head-to-head tests with structure-based models, ProtoBind-Diff holds its own in docking and Boltz-1 (and, spoiler, Boltz-2—data coming soon) benchmarks. It even shines on tough targets with sparse training data.
Here’s the kicker: despite never seeing 3D info during training, its attention maps line up with predicted binding residues. It’s like the model learns spatial interaction rules just from sequence data. This could be a game-changer for ligand discovery across the proteome—especially for orphan, flexible, or new targets where structural data is shaky or nonexistent.
Check the link in the first comment for details (public demo dropping soon). As always, give a follow, like, and repost to keep our spirits high—nothing boosts my ego quite like your attention!
1/2
13
33
227
15,482
Michael Geer retweeted

Link with more info: biopunklab.com/conference
This conference is being organized by my dear friend and notable community builder @ThatMrE (with the amazing team at Cellsius Labs @BiopunkLab), so you can trust that it'll be FUN ⚡️
2
18
1,551
Michael Geer retweeted
Announcing the Biopunk (Un)conference & RAVE: a gathering of biotech rebels and builders 🧬🔧👩🎤
📆 May 2-4th, 2025 in San Francisco
With speakers from Ginkgo Bioworks, NVIDIA, Opentrons, E11 Bio, Pioneer Fund, Lowercarbon Capital, The Odin, Foresight Institute, Tomorrow Bio & more 🎙️
3 days, 3 tracks: open bio (making bio accessible for all), heretical ideas (crazy-sounding-but-feasible) & "crash n burn" (tales of biotech building gone awry) 🤟
Biopunk is not your usual snooze-fest conference – it is a space for open discussion and dissent!
Dive into sessions on: gene hacking, brain mapping, cryopreservation, greening the planet, making bee-free honey, dodging biosecurity disasters, fixing biolab supply chains, chasing eternal life, sperm racing, boosting brains, and tales from biotech’s wild misfires.
All with a renegade edge 👩🎤⚡️
Come prepared with your most controversial (informed) opinions! (and your best biopunk moves for the rave 😎)
@BiopunkLab

8
29
126
19,449
Michael Geer retweeted

15 Jan 2025
On-orbit commissioning is now complete! Up next, Blue Ghost will spend approximately 25 days orbiting Earth 3.5 times at varying altitudes that get us closer to the Moon each time. Check back for daily updates along the way! #BGM1 fireflyspace.com/news/firefl…
35
184
1,137
57,966
Michael Geer retweeted

20 Nov 2024
Joe Rogan blasts the New York Times for their insane fact check on RFK Jr. about Fruit Loops.
“The New York Times just debunked, in the most insane way, RFK Jr’s assertion that the ingredients in Froot Loops are different in Canada than they are in the United States.”
“They're literally saying he was wrong, but he was right.”
“If we're saying that these things have been eliminated in other countries because they've been proven to be dangerous, what is your motivation for saying he was wrong?”
“This left wing rejection of RFK Jr. because now he's connected to Trump, which is connected to Nazis. It's like you go down this weird rabbit hole with these people.”
“Are you trying to remove all leftover credibility because you lost so much credibility already.”
53
776
3,451
134,379
Michael Geer retweeted

19 Nov 2024
Thrilled to receive a scholarship for @athena_DAO_'s upcoming Fertility 101 Course! Looking forward to expanding my knowledge in reproductive health! 🧬🧞♀️

2
2
19
1,025
Michael Geer retweeted

12 Nov 2024
على هامش قمة الرياض العالمية للتقنية الحيوية الطبية،نظمت القمة العديد من الاجتماعات الثنائية بين ممثلي الجهات الحكومية والشركات المحلية والعالمية المشاركة، حيث بلغ عدد هذه الاجتماعات أكثر من 100 لقاء،بمشاركة أكثر من عشر جهات حكومية ذات صلة بمجال التقنية الحيوية الطبية.
#RGMBS2024
2
17
45
22,004
Michael Geer retweeted

16 Aug 2024
Yes, but importantly when AI is trained to make decisions it is trained on how we THINK we make decisions. For example, we think we want to win war by only killing enemy soldiers till they give up, but actually humans rarely fire their weapons at each other and eventually we negotiate a peace and not as many people die. If an AI follows what we say is our objective the death count will skyrocket and wars will escalate not reduce. We need to not give firing decision ability to the AI or we remove the powerful human safety barrier.
1
2
117
Michael Geer retweeted

14 Jul 2024
When we treat our political rivals like they are the enemy of the state, we radicalize our country. I am very glad Trump is safe. The demonization of his image by the DNC and legacy media is responsible for the hysteria that is ripping this country apart. Now having been on the receiving end of the DNC's campaign of lies, I know most of what they've said about "Trump's danger" is an absolute falsehood. My heart breaks for the family of the individual whose life was lost at the rally in such a violent way. Let this be a lesson to all political leaders: when you dehumanize your opponent and radicalize your base, you are responsible for the violence that inevitably ensues.
750
3,881
21,842
1,271,304
Michael Geer retweeted

14 Jul 2024
My full interview with NewsNation
1,042
2,912
18,774
1,547,871
Michael Geer retweeted

14 Jul 2024
A truly presidential and beautifully poetic, compassionate, and inspiring commentary by @RobertKennedyJr on @NewsNation about the attempted assassination of President Trump.
24
181
723
336,465

Michael Geer retweeted

28 May 2024
Live stream of the debate between @aubreydegrey and @fedichev from @foresightinst x.com/i/broadcasts/1gqGvQarz…
4
11
36
4,246
16 May 2024
When did we all start being OK letting political parties decide who voters get to see debate? This should surely be based on average percentages in some set of polls. When did everyone stop caring about actually having democracy? Lovely to see grown men giggling about subverting democracy on live tv.
16 May 2024

The only thing the DNC & RNC agree on is they don’t want @RobertKennedyJr to debate their candidates 😂
1
129
Michael Geer retweeted

16 May 2024
The only thing the DNC & RNC agree on is they don’t want @RobertKennedyJr to debate their candidates 😂
223
404
2,386
151,498
Michael Geer retweeted

24 Mar 2024
Large Biological Models.
This where AI will start to really make a difference in disease and medicine.
When Large Biological Models (LBMs) get up and running and optimized such as we have seen with Chat GPT - disease treatment will change far beyond our current frameworks.
longevity.technology/news/di…
4
22
2,829
1 May 2024
Amazing being the inaugural podcast guest on the already legendary Nina's Notes science podcast. We cover a great overview of how @humanity_app and the healthspan industry are working to make you healthier and happier.
podcasts.apple.com/us/podcas…
3
539
Michael Geer retweeted

3 Apr 2024
🎉We are excited to officially share that SENS Research Foundation and Lifespan.io will be merging, upon completion of regulatory approvals, coming together to form a unique longevity nonprofit.
📰Full PR here:
lifespan.io/sens-lifespan-me…
@senstweet
@LifespanIO

8
9
49
12,609