
Long/short HC with drooly dog. Don't believe me, see for yourself.
Joined January 2021
- Tweets 3,248
- Following 134
- Followers 908
- Likes 15,525
429 Photos and videos











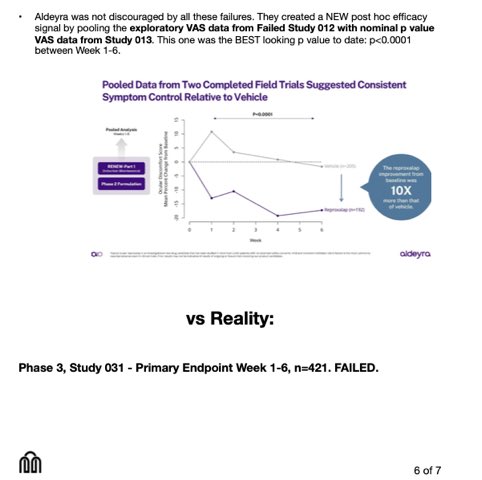


$ABVX — strong framework, but it's using mgmt's background which seems to be inflated. The 0.3–0.7 (non-NMSC) / 0.7–1.4 (NMSC) ranges depend on thiopurine-treated cohorts: the 1.36 NMSC ceiling is literally Abbas year-5 thiopurine, plus a Kaneko "5× prostate" that I couldn't find in the paper.
Thiopurine-naïve UC is ~0.3–0.5 / ~0.4–0.9 → events-to-signal drops well below 10–14.
On NMSC: the ABX464-104 protocol (NCT04023396) discloses the N-Glu metabolite is phototoxic at clinically relevant exposure — a real drug-specific BCC/SCC mechanism.
So skin cancers aren't purely background, even if manageable.
Sources: Abbas 2014 → pubmed.ncbi.nlm.nih.gov/2524… 2024 → pubmed.ncbi.nlm.nih.gov/3859… (NCT04023396) → cdn.clinicaltrials.gov/large…

Based on our conversations with the buy side and sell side, one thing is clear: the market is still struggling to define the actual safety bar for $ABVX.
Specifically: how many non-NMSC events would ABTECT Maintenance Part 2 need to show before FDA reviewers or sophisticated pharma BD teams should become concerned that obefazimod may have a true carcinogenic signal, rather than background malignancy events in a refractory UC population?
We provide our analysis and conclusions below. Said simply: under our assumptions, $ABVX would need to show a malignancy burden far beyond one or two incremental cancer cases before the bear case becomes statistically and biologically coherent.
When a drug trial reports a handful of cancer cases, the natural question is: is this a real signal, or just bad luck? A drug study with 150 patients over one year will almost certainly see some cancers — because UC patients get cancer at a background rate even without any drug. In fact, even otherwise healthy people get cancer at a background rate without any drug. The real question is whether the number of cancers seen on the drug is more than we would expect by chance from that background rate alone.
Enter statistics: the formal language of separating signal from bad luck. In this case: what is the event burden and pattern that is high enough, coherent enough, and biologically plausible enough to overcome a low causal prior to obefazimod being carcinogenic?
This is exactly the kind of question Bayesian inference is useful for answering.
Our Bayesian inference model asks a simple question:
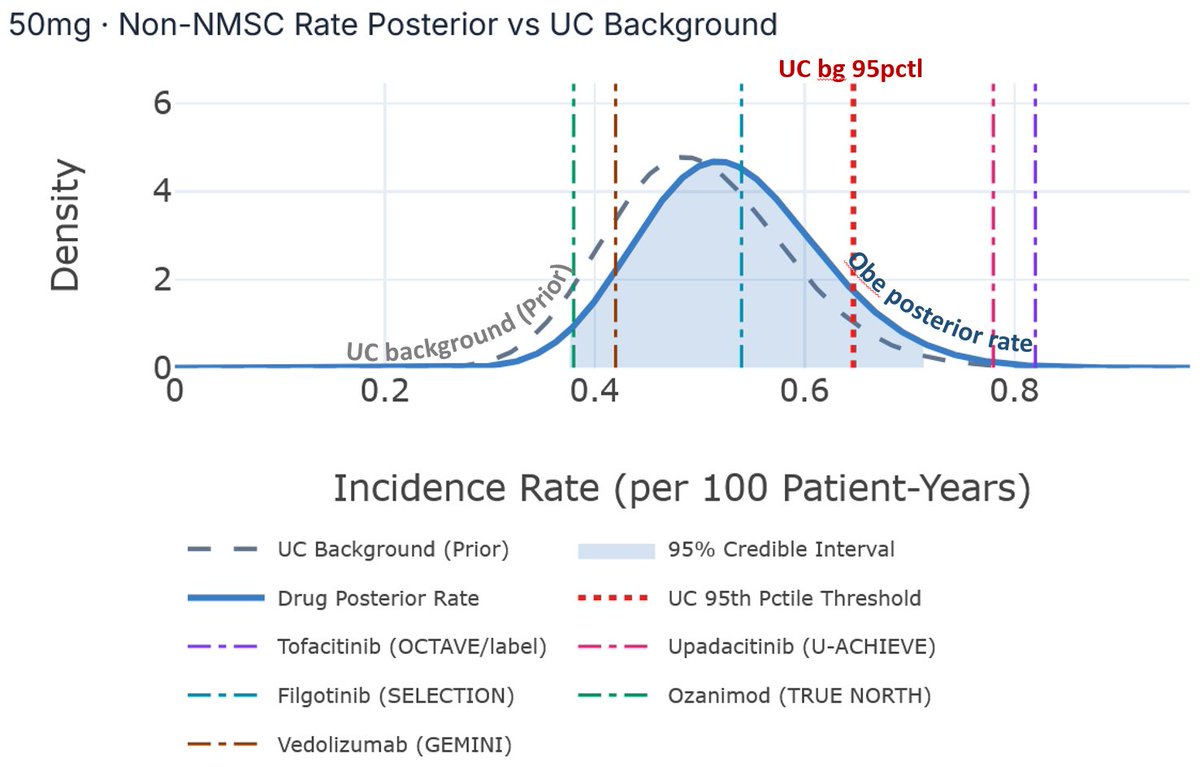
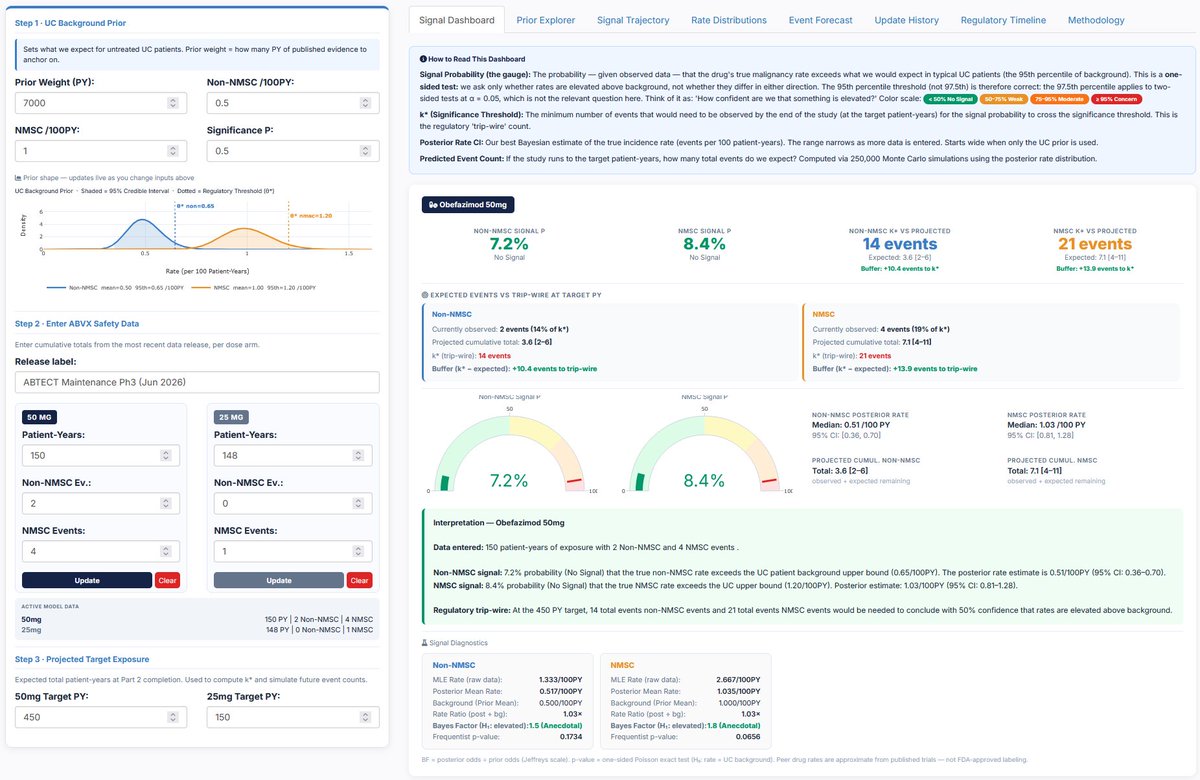
Given an estimated background non-NMSC incidence rate of ~0.5/100 PY, how many additional non-NMSC events would need to appear in the next ~450 PY of 50 mg exposure before the pooled 50 mg maintenance dataset begins to look meaningfully above UC background?
At ~0.5/100 PY, the expected background incidence over ~450 PY is roughly 2–3 non-NMSC events.
Our model suggests the upcoming 50 mg Part 2 update would need to show something closer to 10 additional events above background — roughly 14 total non-NMSC events in the pooled ~600 PY 50 mg maintenance dataset — before the posterior begins to suggest a >50% probability that the true obefazimod non-NMSC rate exceeds the upper bound of UC background.
We favor a Bayesian inference framework in these cases because it contemplates the data through a lens similar to that of what we believe an FDA reviewer and pharma regulatory teams would look through: exposure-adjusted rates, confidence intervals around those rates, event heterogeneity, organ clustering, baseline UC cancer risk, prior therapy risk, timing of exposure, investigator attribution, and whether the pattern resembles a broader immunosuppressive AE phenotype.
Most importantly, with this approach we do not need to assume obefazimod causes cancer. In fact, the causal prior should be low because there is no obvious a priori hypothesis for obefazimod causing cancer, including clean preclinical work, no clustering to a specific cancer type, no broader adverse event pattern associated with impaired immune surveillance, and investigator assessment that the disclosed prostate and breast cancer cases were unlikely related.
Thus, the analysis is not whether “observed cancers divided by patient-years is greater than zero.”; that is coarser than the data allows. The question is whether the observed event pattern is adequate to overcome a low causal prior.
We estimated background malignancy IRs across an aggregate 7000 PY based on a synthesis of Colombel et al. (2017), Rubin et al. (2026), D’Haens et al. (2023), Swissmedic (2024), Sands et al. 2026, Abreu et al. (2022), and Sandborn et al. (2019), and arrived at an estimated non-NMSC IR of 0.5, which is the midpoint of the range provided by management in the Part 2 primer deck published earlier today (ir.abivax.com/static-files/7…). We use 0.5/100 PY as the non-NMSC IR as the background prior of our model. Our analysis allows us to use 7000 PY as the weight of that prior probability. Then, we ask how many non-NMSC events need to be observed in the ~450 PY from Part 2 for the aggregate obefazimod non-NMSC IR to indicate that the true obefazimod non-NMSC rate exceeds the UC patient background upper bound with at least 50% probability. We set the PY reported in ABTECT Maintenance Part 1 according to management feedback.
An estimated background non-NMSC IR prior of 0.5/100 PY is at the midpoint of the range provided by $ABVX management and implies an expected 2-3 non-NMSC events per 450 PY. Our model suggests that the ABTECT Maintenance Part 2 update for the 50 mg dose would need to show 10 additional events over and above those background events in 450 PY (i.e., total of 14 non-NMSC events) before the pooled 600 PY maintenance dataset (Part 1 Part 2) for the 50 mg dose to indicate a greater than 50% probability that the true obefazimod non-NMSC rate is greater than the upper bound of UC background rate, when the observed non-NMSC IR begins to possibly be considered elevated above background expectations.
Even when we cut the prior probability weight in half to 3500 PY, the conclusions remain unchanged: ABTECT Maintenance Part 2 update for the 50 mg dose would need to show 7 additional non-NMSC events over and above the number of events expected given the background IR before the pooled 600 PY maintenance dataset (Part 1 Part 2) for the 50 mg dose to indicate a greater than 50% probability that the true obefazimod non-NMSC rate is greater than the upper bound of UC background rate.
Said differently, the key risk is not that another cancer event occurs; the UC and IBD literature documents an expected incident rate for non-NMSC at their background rate (0.3-0.7/100 PY).
The key question is whether the next ~450 PY expected from the Part 2 50 mg arm is consistent with the drug-driven malignancy hypothesis: repeated non-NMSC events, organ clustering, biological patterning, exposure-duration logic, or a broader immune-surveillance signal.
That is what our model tests.
If the next cut instead shows low-count, disparate, background-plausible events, the current obefazimod-driven malignancy argument becomes increasingly difficult to sustain.
But what about Rinvoq? They didn’t see a big imbalance?
This is not a Rinvoq/Xeljanz-style RA safety signal, where a nearly 4,500-patient outcomes study produced a statistically significant malignancy and immunosuppressive AE cluster in line with an a priori safety hypothesis prior. So far, these few malignancy observations are easily explainable by the background rate of UC cancer in this population especially, and mechanistically, there is also no obvious reason to believe obefazimod should be carcinogenic. If anything, Qin et al. (miRNA-124 in Immune System and Immune Disorders - PMC) discuss miR-124 biology in anti-proliferative and anti-fibrotic contexts, though we would not underwrite an anti-cancer effect here.
Against the magnitude of efficacy demonstrated by obefazimod to date: ~50% clinical remission versus ~10% placebo, plus large effects on endoscopic remission, HEMI, steroid-free remission, and sustained remission that is life-altering; these non-NMSC events do not appear to alter the risk-benefit equation.
We do not think these disclosed data support a black box. A black box for what appears likely to be background artifact would risk causing net harm to patients by creating unnecessary friction around delivery of a novel and highly efficacious therapy.
A brief comment on NMSCs: Based on our diligence, including conversations with former FDA review leadership responsible for evaluating new IBD drug approvals, NMSCs are most commonly basal cell carcinoma and cutaneous squamous cell carcinoma. Importantly, these cancers are: 1) very common; 2) very slow growing; 3) very visible; and as a result, 4) very treatable and curable. These cancers are rarely metastatic, and very rarely fatal.
There is published evidence that IBD patients appear to be at elevated rates of NMSCs, hypothesized to result from either higher baseline persistent inflammatory processes and/or thiopurine use.
In fact, clinical guidelines published in April 2025 following an expert panel from the American Gastroenterological Association Clinical Practice Update provided specific guidance that all adult patients with IBD should follow skin cancer primary prevention practices by avoiding excessive UV exposure, that patients on immunomodulators, anti-TNF biologic agents, or small molecules should undergo yearly total-body skin exam, and that patients with any history of thiopurine use should continue yearly total-body skin exam even after thiopurine cessation.
In the cohort study, the incidence of NMSC was higher among patients with IBD compared to controls (IRR 1.64, 95% CI 1.51–1.78). Recent thiopurine use was associated with NMSC (adjusted OR 3.56, 95% CI 2.81–4.50), and persistent thiopurine use was associated with NMSC (adjusted OR 4.27, 95% CI 3.08–5.92).
While, prior to our analysis, we were not concerned that the FDA or pharma would view elevated NMSC rates as a major issue, we were concerned that investors might continue to misclassify them as a serious systemic malignancy signal. However, based on recent conversations with other buysiders, we sense the market is beginning to understand that NMSCs are not a meaningful issue here: they are generally detectable, manageable, and already incorporated into the standard-of-care dermatologic monitoring framework physicians use for UC patients.
For completeness, we repeat our Bayesian inference analysis as described above but for NMSC. Taking the NMSC background IR of 1/100 PY provided by management today, we expect 4.5 cases in a 450 PY dataset. Our model suggests that the ABTECT Maintenance Part 2 update for the 50 mg dose would need to show 12 to 13 additional events over and above those background events in 450 PY (i.e., total of 21 NMSC events) before the pooled 600 PY maintenance dataset (Part 1 Part 2) for the 50 mg dose to indicate a greater than 50% probability that the true obefazimod NMSC rate is greater than the upper bound of UC background rate, when the observed NMSC IR begins to possibly be considered elevated above background expectations.
In summary: The right conclusion is not that no cancer events occurred, or that no events will occur in the future. They will occur, not only because cancer risk is elevated in UC, but because cancer can occur in anyone, at any time, including otherwise healthy patients.
We think the right conclusion is that the events that did occur look explainable by baseline patient risk, common cancer epidemiology, UC biology, and routine screening detection, rather than acting as evidence of an obefazimod-specific oncogenic signal.
We think FDA reviewers and pharma regulatory teams are more likely to reach this conclusion than to view the disclosed cases as a coherent obefazimod-specific malignancy signal, making increasingly clear what we believe is a best-in-disease efficacy/risk profile in the lucrative maintenance setting for UC and substantially de-risking the company’s opportunity in Crohn’s.
Disclosure: I/we may be long ABVX and may buy, sell, hedge, or otherwise change exposure at any time without notice. Not investment advice or a recommendation. Analysis reflects current views and assumptions based on public disclosures, published literature, and non-confidential conversations, and may change as new data become available. No compensation from ABVX or any third party.
4
22
8,345

May 26
People forget so quickly….
$SRRK is just as confident in their “accelerated timelines” at the new facility as with all the progress and “FDA alignment” about Catalent.
Given what FDA told $NVO in February, what “alignment” was Hallal talking about in re-inspecting Catalent? FDA was not in a rush to re-inspect, because as they communicated to $NVO in January and February, the site wasn’t ready for reinspection.
May 25

$SRRK 🔫🦶🤦♂️
I think $SRRK rushed BLA re-filing triggered pre-mature FDA re-inspection of Catalent:
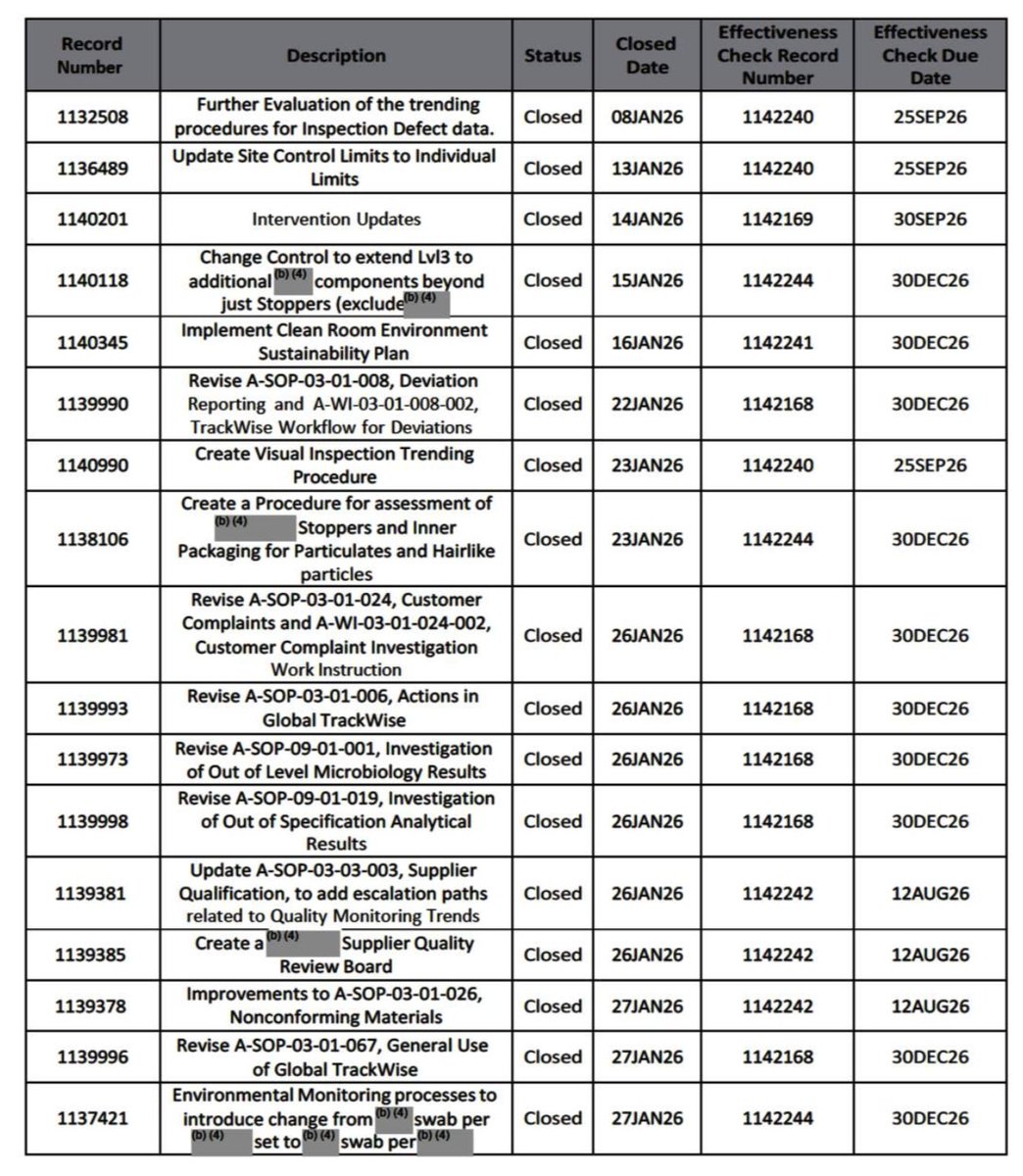
-On February 20, 2026 FDA informed $NVO that re-inspection would follow CAPA effectiveness data (below).
-This data was only due in December 2026 for 26 out of 40 CAPAs, as you can see from a May 7, 2026 letter NVO sent to FDA. (below)
=Even though it looks like FDA had no intention of re-inspecting Catalent before they made sure CAPAs worked, $SRRK forced the FDA's hand:
=the US BLA re-filing required the inspection,
=refused to withdraw the EU filing, even though that has no hope of approval with an OAI and Warning Letter, and the upcoming decision in EU also required the re-inspection.
=so the FDA didn't waste time and re-inspected...
Full text of the $NVO and $SRRK correspondence: tinyurl.com/NVOandFDAdocs
7
3,988
May 25
$SRRK 🔫🦶🤦♂️
I think $SRRK rushed BLA re-filing triggered pre-mature FDA re-inspection of Catalent:
-On February 20, 2026 FDA informed $NVO that re-inspection would follow CAPA effectiveness data (below).
-This data was only due in December 2026 for 26 out of 40 CAPAs, as you can see from a May 7, 2026 letter NVO sent to FDA. (below)
=Even though it looks like FDA had no intention of re-inspecting Catalent before they made sure CAPAs worked, $SRRK forced the FDA's hand:
=the US BLA re-filing required the inspection,
=refused to withdraw the EU filing, even though that has no hope of approval with an OAI and Warning Letter, and the upcoming decision in EU also required the re-inspection.
=so the FDA didn't waste time and re-inspected...
Full text of the $NVO and $SRRK correspondence: tinyurl.com/NVOandFDAdocs
1
1
9
6,538
May 24
$SRRK is 💩 on their shareholders, lol!
Catalent re-inspection = TOTAL FAILURE!
No - Warning Letter closeout in 2026
No - EU approval
No - Early FDA approval
No - Mgmt credibility
Read the Form 483: tinyurl.com/NVO2026Form483

7
19
9,561
Apr 1
$SRRK
Here is the summary of the Jan 14 FDA meeting with Novo: tinyurl.com/NVOmtgJan14
-FDA disagrees with Novo conclusions
-FDA questions reliability of their data
-FDA asked “critical” questions which Novo FAILED
FDA wrote this summary, not some short seller.
SRRK painted a beautiful picture talking about THIS meeting on their calls. FDA didn’t raise new questions, everyone’s aligned, blah blah blah
I don’t know what the FDA will, or will not do, but what I do know is that $SRRK mgmt has not been accurate in describing Novo’s interactions with the FDA so far.
…I have been pretty good about getting relevant documents here. I was the first one to get the Form 483, the OAI decisional letter AND this meeting summary.
…however, I do not believe there is any record of any March FDA visit to Catalent 🤷♂️
4
1,274
Apr 1
$SRRK
This was Novo’s response to the Warning Letter, submitted on December 12: tinyurl.com/NovoWLResponseDe…
-limited third-party reviews
-CAPA’s still not effective
-tons of plans and new SOP’s
-zero validation data showing effectiveness
-mostly procedural fixes, limited structural changes
-etc
So FDA convened another regulatory meeting on January 14, 2026
3
901
Apr 1
$SRRK
Only after this meeting did Novo start to take things seriously.
Here is their October 3, 2025 response to the regulatory meeting: tinyurl.com/NovoOct3Response
-They admitted that contaminated product had been released.
-They finally classified the cat hair as “extrinsic”
-Started to hunt for a definitive root cause (which they didn’t identify as of January 14, 2026)
Since this was too little, too late, FDA issued a Warning Letter.
2
768
Apr 1
$SRRK
Novo responded to the Request for Additional Information on Sep 5. tinyurl.com/NVOsep5Response
They still didn’t get it. They tried to frame cat hairs as “intrinsic” particles, instead of as extrinsic contaminants.
FDA was not happy with this response either and had to conduct a regulatory meeting on September 26, 2025.
5
918
Apr 1
$SRRK management wants full transparency.
Let’s start at the beginning…
Here is the so-called “comprehensive” response to the Form 483 submitted by Novo on Aug 4, where they:
-blamed issues on legacy Catalent “rapid growth”
-insisted that 100% visual inspection to catch cat hairs was sufficient
-etc
FDA obviously didn’t agree with this an issued a Request for Additional Information on August 27.
RAI are used when responses are insufficient.
At the time $SRRK argued that this response would resolve the 483 and they expected to receive approval. That was insane.
It’s a large file: tinyurl.com/Novo483Response
5
972