
No longer on Twitter, we can keep in touch here: fediscience.org/@cvanderaa
Joined January 2020
- Tweets 126
- Following 211
- Followers 173
- Likes 225
28 Photos and videos




20 Dec 2023
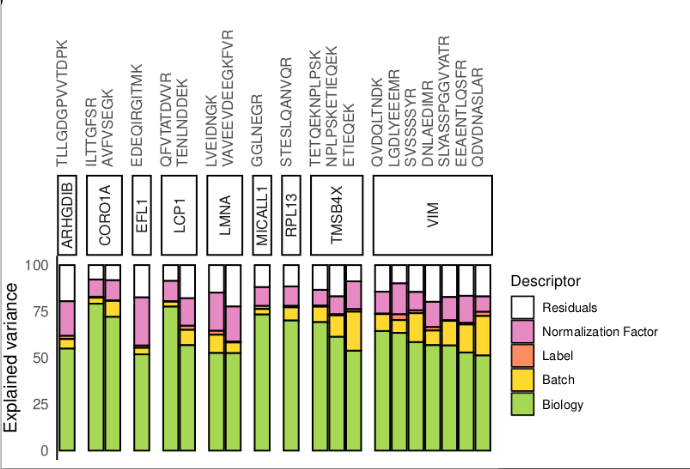
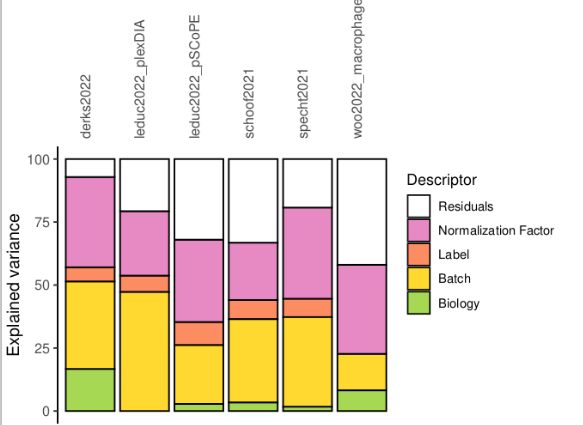
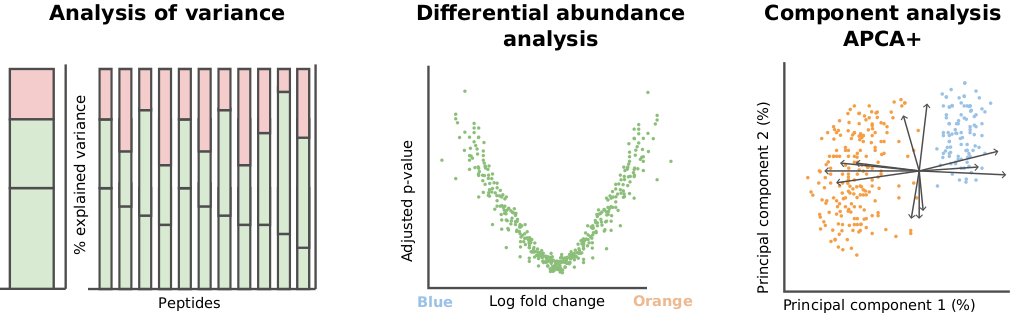
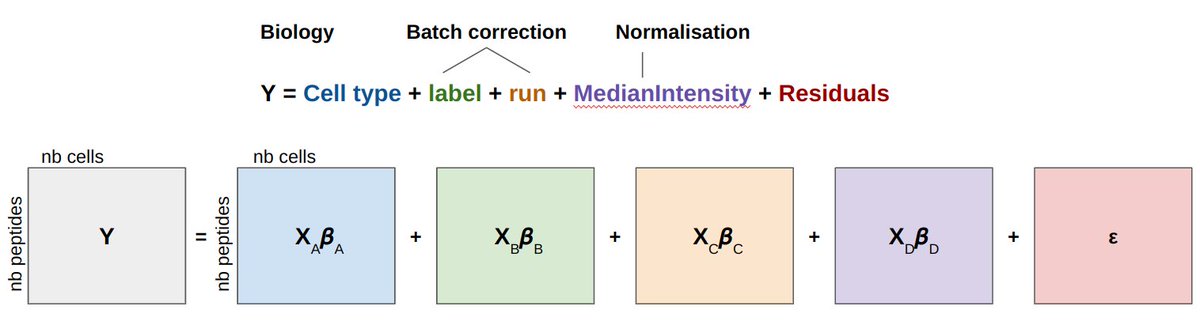
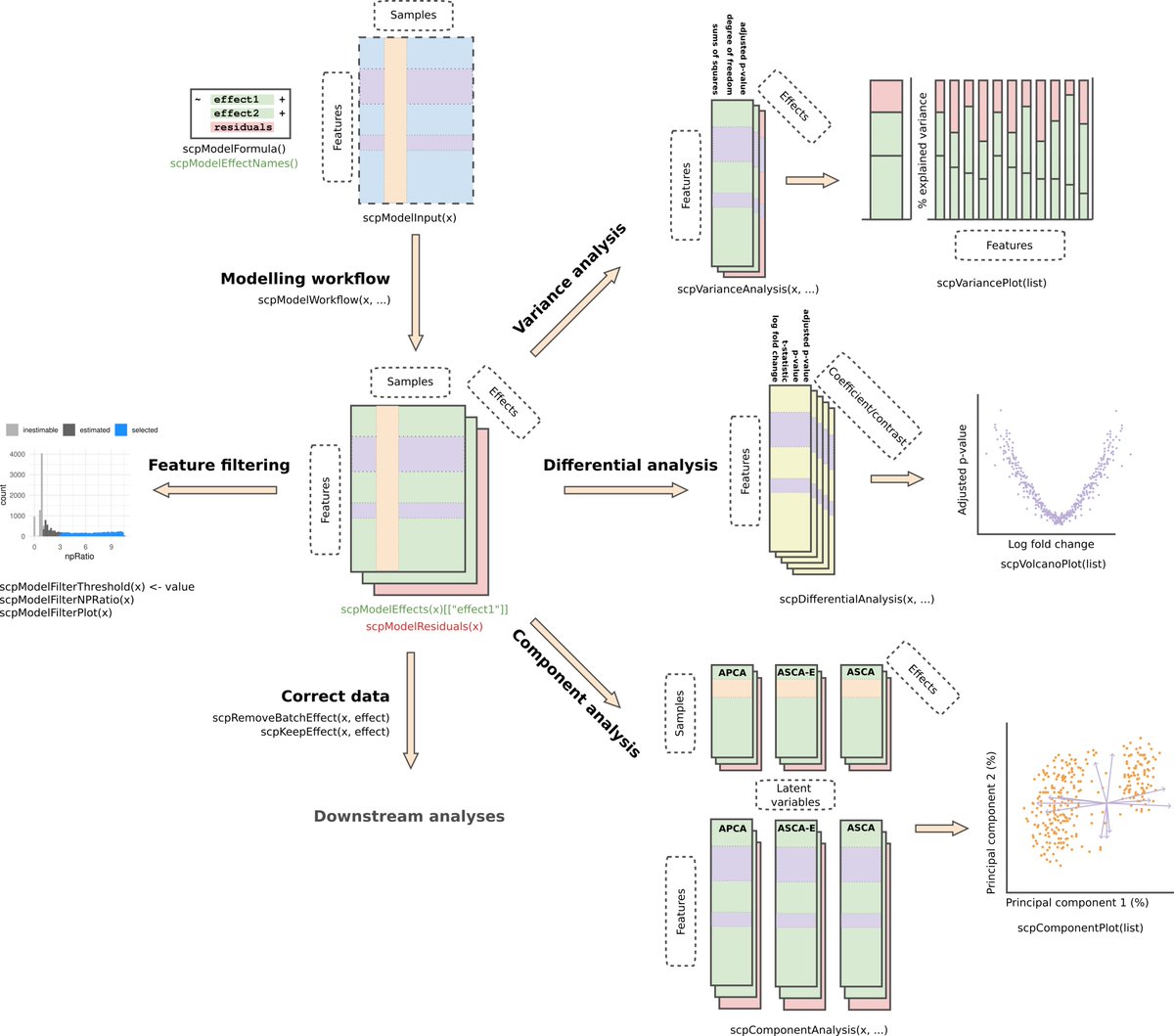
Our new preprint is available on biorXiv: doi.org/10.1101/2023.12.14.5… !🥳 We present scplainer, a principled computational approach to streamline and standardise #MassSpec #SingleCell #proteomics data analysis. Here's a 🧵 with our key messages.
1
15
48
8,065
20 Dec 2023
scplainer will be (very) soon available in scp so that you can try it
yourself. Need data? All data used in this work were retrieved using
our scpdata package!
github.com/UCLouvain-CBIO/sc… github.com/UCLouvain-CBIO/sc…
1
1
146
20 Dec 2023
As always, many thanks to my promoter Laurent Gatto for his supervision. I also warmly thank Manon Martin and Bernadette Govaerts for their guidance on the APCA framework.
207
Christophe Vanderaa retweeted

14 Sep 2023
Our last work on #SingleCellMultiModal data collection is finally out! 🎉
It has been a long journey 🚀, but it taught me a lot! 🥸
Thanks to all the co-authors (in the comments), I really enjoyed working with all of you! 🤩
doi.org/10.1371/journal.pcbi…
(1/5)
2
20
72
13,309
20 Apr 2023
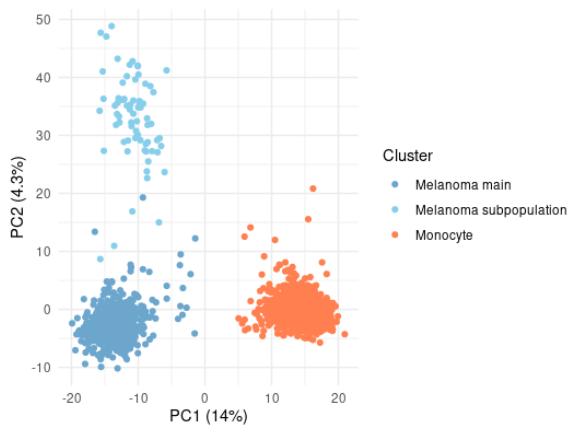
And I forgot to mention that we performed data exploration and illustration using our @Bioconductor tools: scp and scpdata 😅

14 Apr 2023

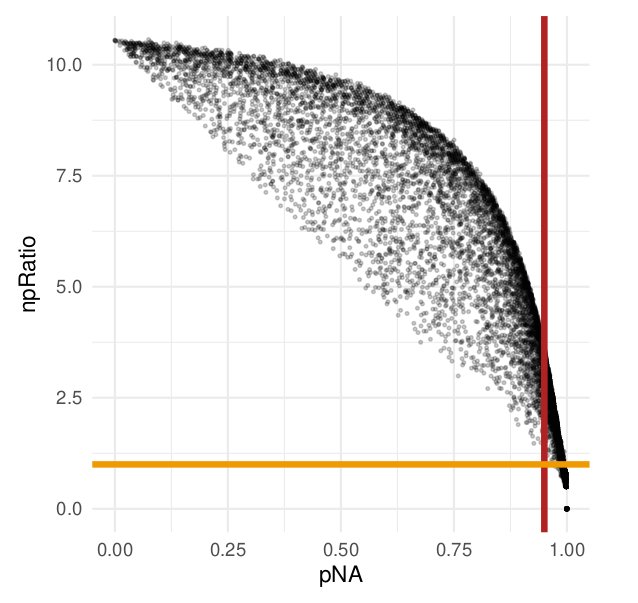
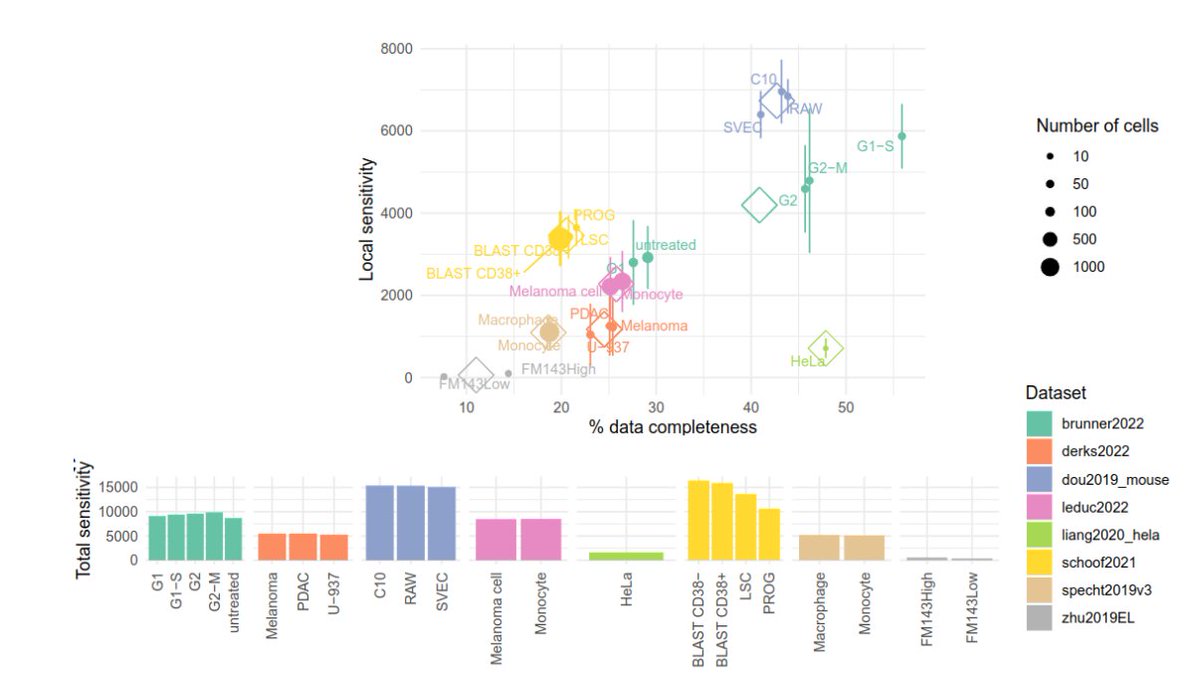
So glad to share with you our new preprint: arxiv.org/abs/2304.06654 🥳 In this perspective, we discuss how to deal with missing values in #MassSpec #SingleCell #proteomics. Here's a 🧵 with our key messages.
7
472
14 Apr 2023
#Reproducible data analysis is so important! Our decision to emphasize on software details and providing code was sparked by an email I received from @harrisonspecht "Reanalysis of reanalysis".
14 Apr 2023
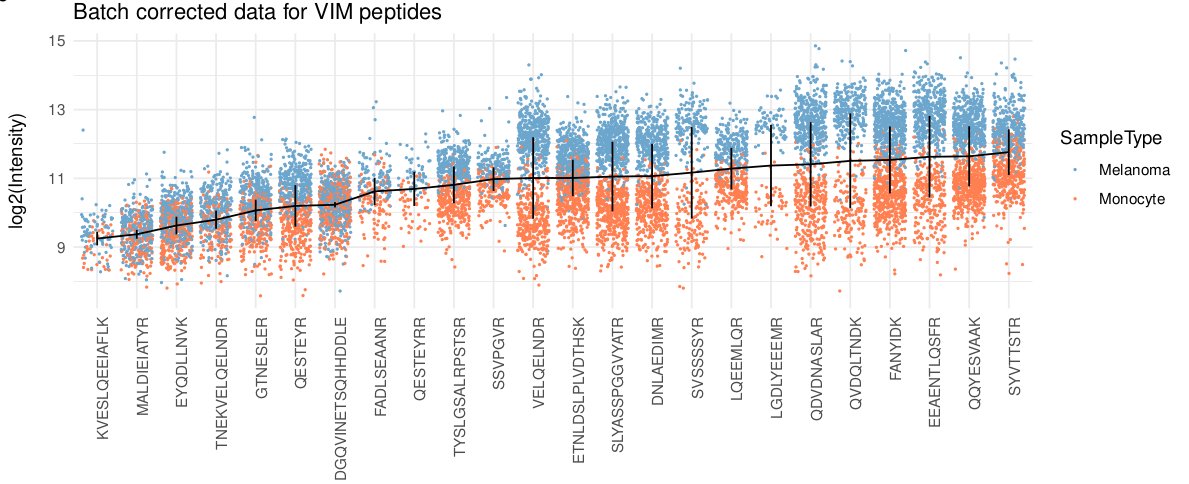
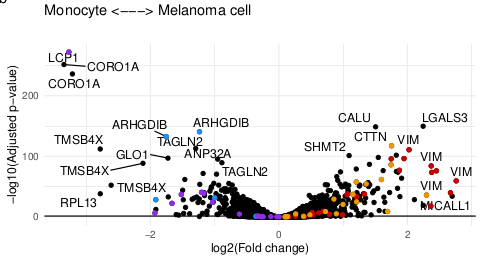
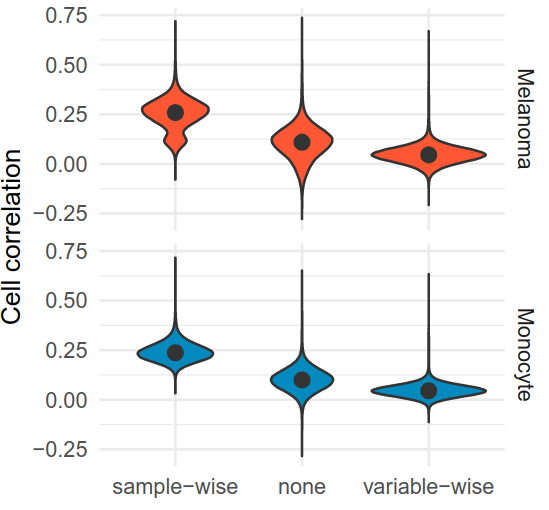
Software details do matter! For instance, the same KNN algorithm implemented in two pieces of software leads to different results, because they either impute by samples (cells) or by variable (proteins). Here's the impact on cell correlations.
1
1
9
849
14 Apr 2023
Harrison raised the points that the correlations he computed were not the same as the correlations we showed in our previous article, while we were both processing the exact same dataset with the exact same algorithms (or were we?) !
1
112
14 Apr 2023
Because the @slavovLab does such a nice job of providing the code to analyze their data, I could quickly identify that we were KNN imputing by variable and Harrison was KNN imputing by sample. This would have taken me ages if I add to guess this from a blurry methods section!
109