
12 Photos and videos









Pranshu retweeted

Jun 11
74
241
4,210
564,131

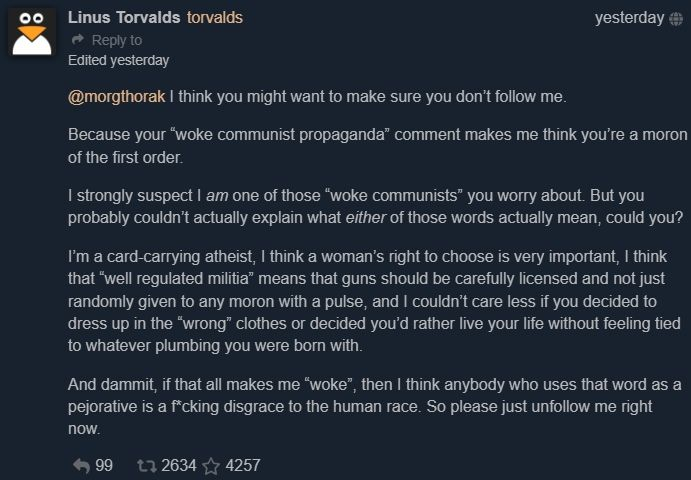
He thinks he's the dev from ready player one 🤣😂
Apr 20

Check out this very goofy, very pathetic man who’s the highest paid CEO on the planet, and thinks that the world should be ruled by a techno-fascist elite that keeps society in a perpetual state of war.
32
Pranshu retweeted

Apr 18
This is the gayest thing I've ever seen, and I've had sex with men.
782
501
11,071
2,614,767
Pranshu retweeted

Apr 16
How Americans spend their day
🎥: EEAGLI
39
379
2,989
489,053
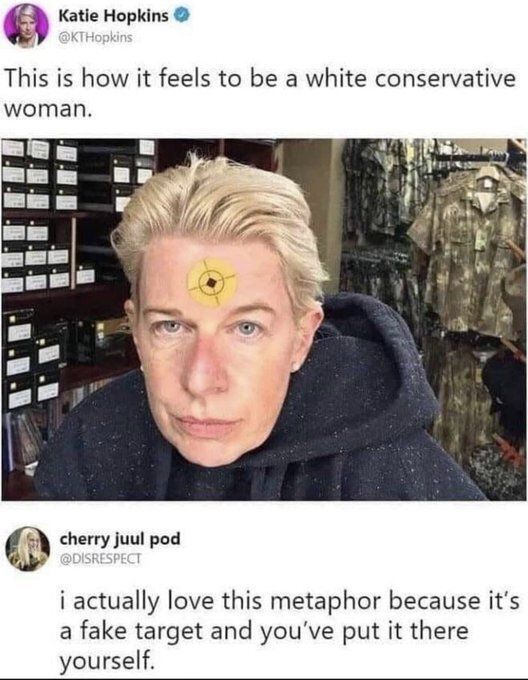
Congrats to him :) but with that hairline, he defo needs to be checked properly for roids

18-year-old GOUT GOUT 🇦🇺 19.67s (1.7) over 200m at Australian Championships in Sydney!!🤯🤯
A new U20 World Record ☑️
National Record ☑️
First Australian man under 20 seconds ☑️
A star is born!
56

Pranshu retweeted

20 Oct 2025
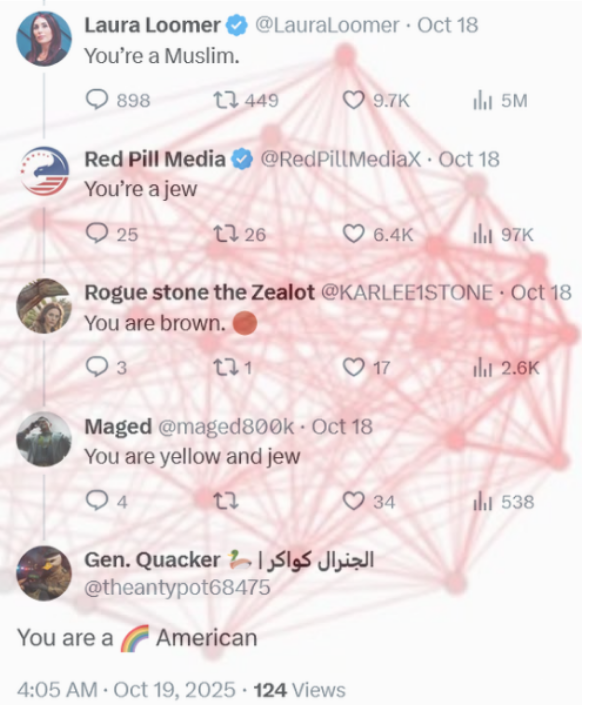
here you go buddy give it a day or so and post this with the same title
2
42
1,395
41,356

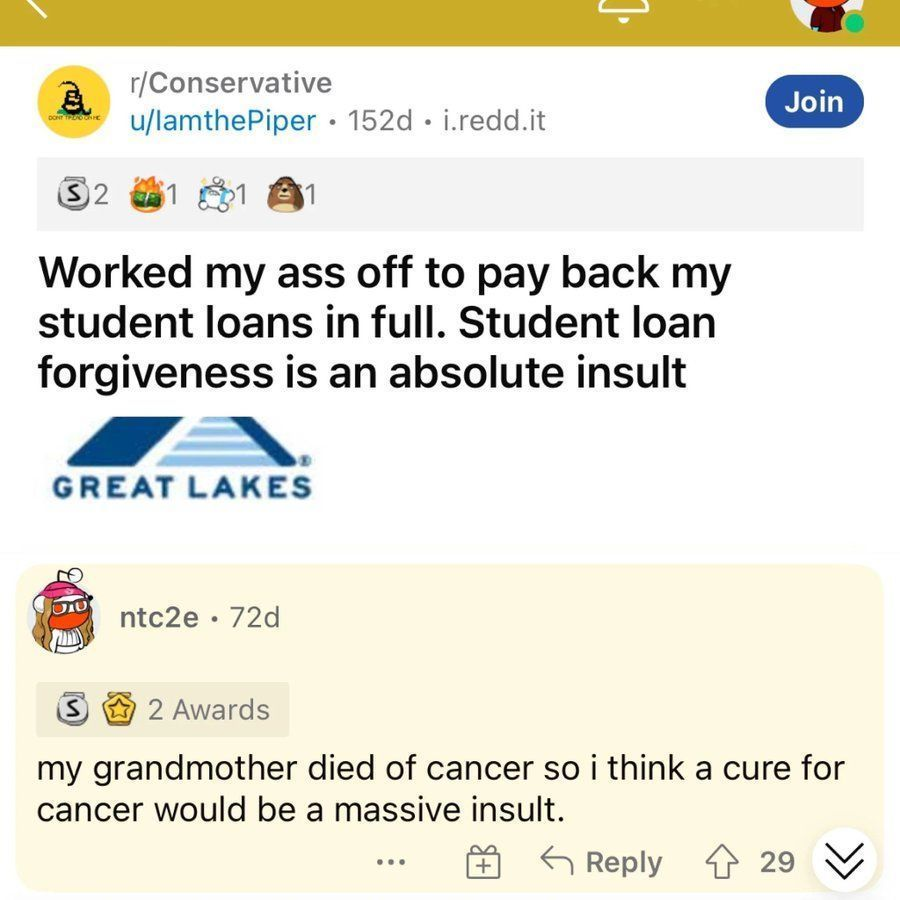
For those who haven't, please read Born a Crime by trevor noah


117






Pranshu retweeted

Mar 17
what's stopping you from coding like this?

354
37
558
31,724
Pranshu retweeted

Mar 14
That techbro knew that highlighting ChatGPT/ Alphafold will make it go viral - Catnip for X bros trying to monetize..
Bet interviewing the researcher involved will reveal how it was done
Per Grok -
### How mutations are usually identified from sequencing data (standard pipeline for neoantigens in personalized cancer vaccines)
The typical workflow for turning tumor sequencing into potential neoantigens (mutated peptides that could be vaccine targets) is a multi-step bioinformatics pipeline. This is done long before any structure prediction tool like AlphaFold comes into play. Here's the usual process in 2025–2026 clinical/research settings:
1. Sequencing:
- Whole-exome sequencing (WES), whole-genome sequencing (WGS), or targeted panels of tumor DNA.
- Matched normal (germline) DNA from blood/saliva for comparison.
- Often paired with tumor RNA-seq (to check expression of mutated genes).
2. Read alignment & pre-processing:
- Align raw reads to the human reference genome (e.g., using BWA, Bowtie2).
- Quality filtering, duplicate removal, etc.
3. Somatic mutation calling / variant detection (this is where mutations are actually identified):
- Compare tumor vs. normal to find somatic (tumor-specific) variants.
- Tools: Mutect2 (GATK), VarScan, Strelka2, MuTect, or ensemble callers for higher confidence.
- Focus on nonsynonymous mutations (missense, indels, frameshifts, fusions) that change the protein sequence.
- Filter for high variant allele frequency (VAF), expression in RNA-seq, clonal status, etc.
4. Translation to mutated peptides:
- Annotate variants with tools like VEP (Variant Effect Predictor) or ANNOVAR.
- Generate the altered protein sequence around the mutation (e.g., 21-mer or sliding windows).
- Split into possible epitope lengths (8–11 mers for MHC class I, longer for class II).
5. Neoantigen prediction & prioritization:
- Peptide-MHC binding affinity prediction (core step): NetMHCpan, MHCflurry, MixMHCpred → rank how well mutated peptides bind patient's HLA alleles (typed via sequencing or PCR).
- Additional filters: proteasomal cleavage (NetChop), TAP transport, expression level, clonality, dissimilarity to self (to avoid tolerance), immunogenicity scores.
- Pipelines: pVACtools, MuPeXI, nextNEOπ, NeoPredPipe, TruNeo, or commercial ones like those from BioNTech/Moderna/Genentech.
Only after this do you have a shortlist of candidate neoantigens (mutated peptides).
### Where AlphaFold actually fits in (limited role)
AlphaFold enters much later — and rarely as the main tool for pinpointing neoantigens:
- To model the 3D structure of a mutated protein or peptide-MHC complex → assess if the mutation dramatically alters folding, stability, or epitope presentation/conformation.
- To help understand why a neoantigen might be immunogenic (e.g., structural changes exposing new surfaces).
- In advanced vaccine design: refine peptide structures for better MHC binding or TCR interaction modeling (e.g., with tools like AlphaFold-Multimer or RoseTTAFold).
- In some emerging pipelines (post-2024): use AlphaFold structures as input features for improved immunogenicity predictors or to simulate neoantigen presentation.
But AlphaFold cannot replace variant calling or read sequencing data — it starts from an already-known mutated amino acid sequence. If someone claims "AlphaFold pinpointed the mutated proteins from genomic sequences," it's either loose wording (they mean used AlphaFold downstream after variant calling) or a misunderstanding of the tools.
In short: sequencing variant callers find the mutations → neoantigen pipelines predict peptides → AlphaFold (optionally) models structures for refinement. The heavy lifting for "identifying mutations" is done by dedicated genomic tools, not AlphaFold.
2
1
7
4,736
Pranshu retweeted

Mar 14
Yup. These cartoon speak volumes about their blind, sycophantic credentialism.

12
2
328
9,472
Pranshu retweeted

Mar 14
This meme was originally meant as pejorative satire and ended up describing uncomfortable reality.






194
2,349
25,909
1,222,093

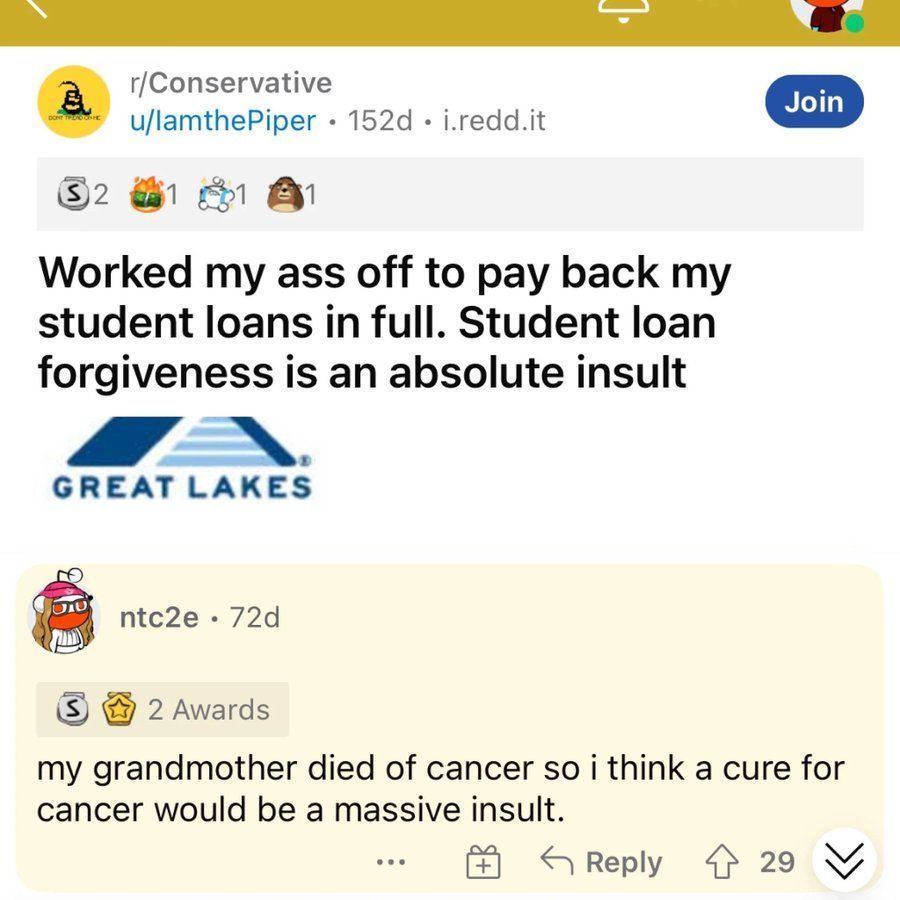
Pranshu retweeted

Mar 11
Atheists are now DEEP FRYING ABORTED EMBRYOS and dipping them in hot sauce.
Why is this not FRONT PAGE NEWS?

1,181
864
5,767
449,800