
Genomics technologist and bioinformatics hacker at the WEHI Advanced Genomics Facility. Views and opinions are my own.
Joined August 2013
- Tweets 1,391
- Following 337
- Followers 393
- Likes 682
14 Photos and videos

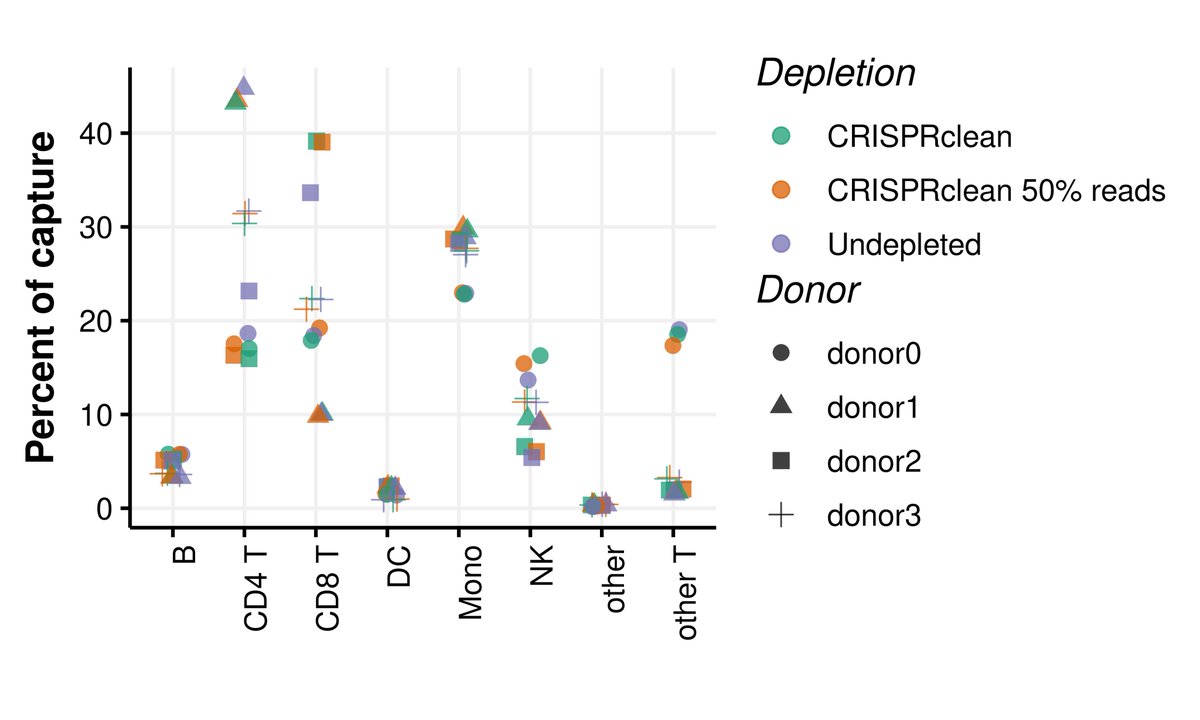
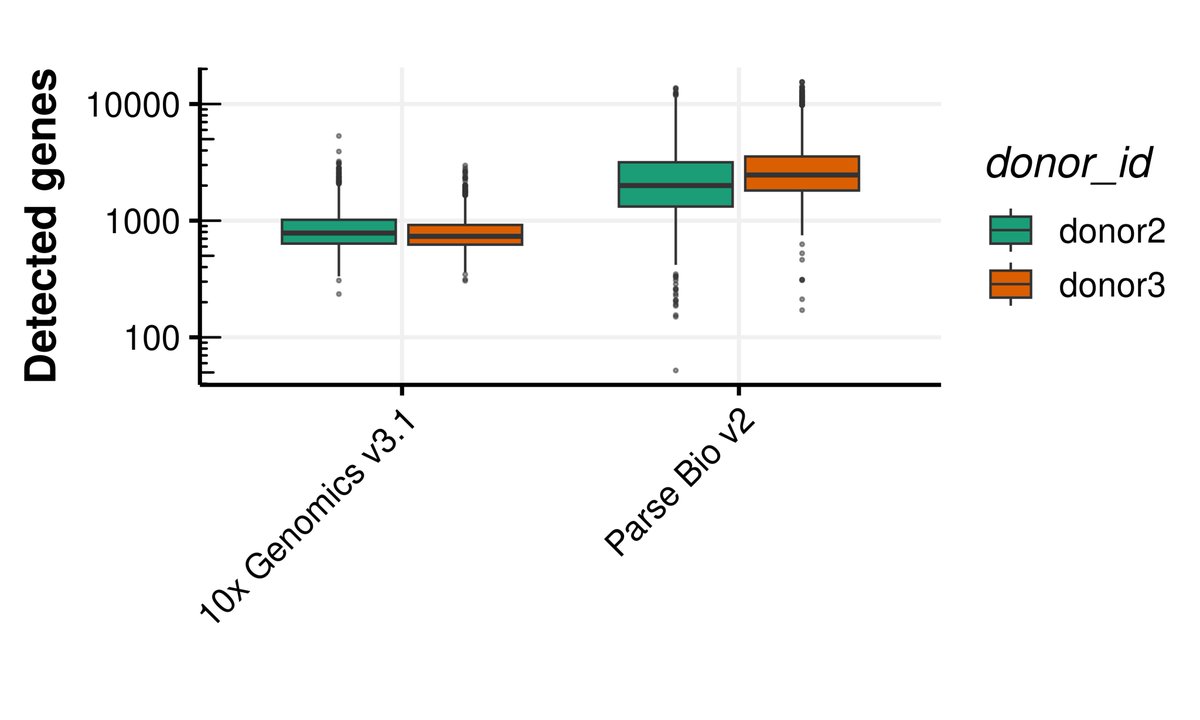
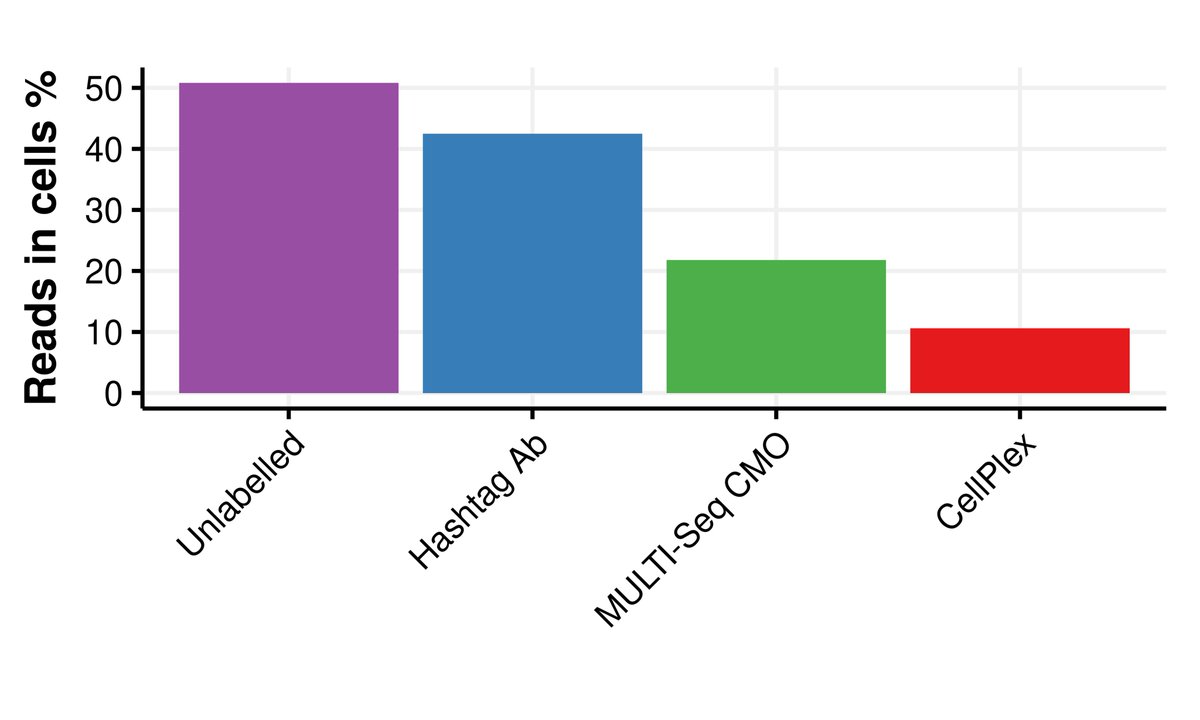




Pinned Tweet

25 Nov 2024
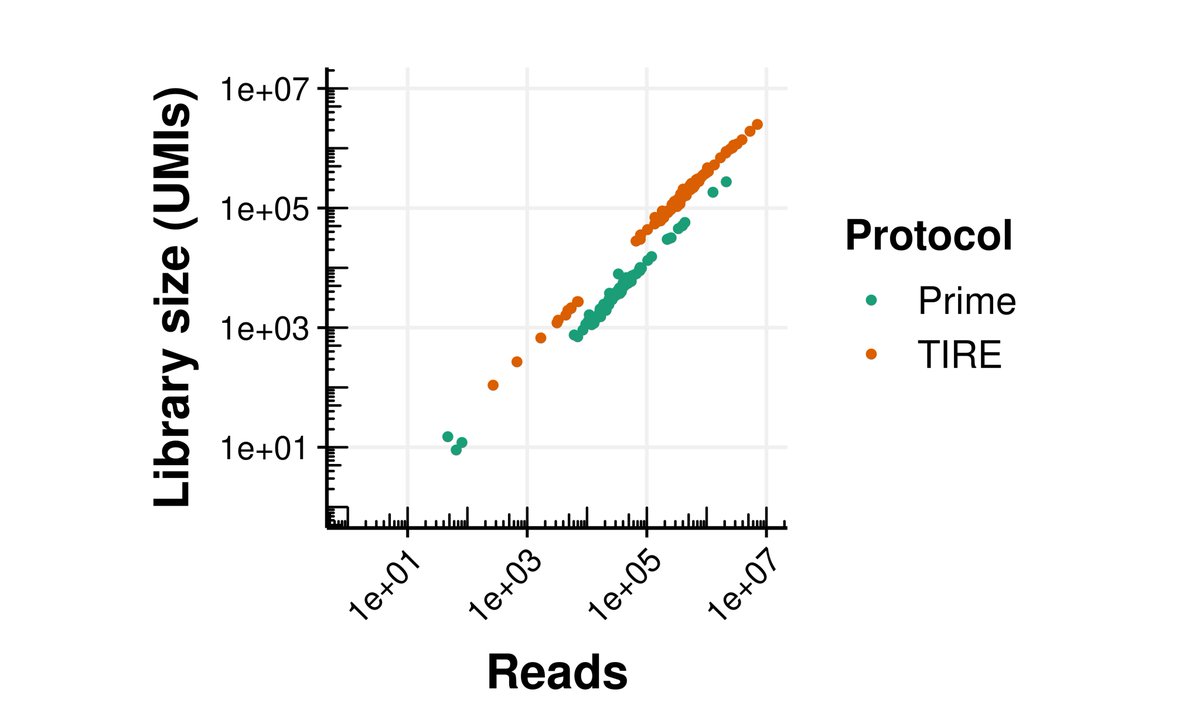
Tired of tedious RNA extractions for your transcriptomic studies? Introducing TIRE-seq, a novel protocol that streamlines bulk RNA-seq by integrating sample extraction with library preparation. The whole process can be completed in a day and a half. (1/6) biorxiv.org/content/10.1101/…
1
2
6
551
Daniel V Brown retweeted

Jun 9
Today we release Rhaister, an elegant statistical model that predicts drug phenotypes in new contexts w/ accuracies comparable to experimental assays.
And dropping Emerald Bay, a 2M cell dataset measuring long time-course phenotypes across 1000s of drug-cell line interactions.
14
54
251
58,117
Daniel V Brown retweeted

Jun 11
Came across an interesting paper today which trains sparse autoencoders on AF3 vs Boltz-2 denoising trajectory.
AF3 builds bottom-up from local physicochemical features residue identity, charge, hydrophobicity early, then global stuff like coiled-coils and tertiary packing etc. whereas Boltz-2 commits to protein-identity and domain features early (pattern matching).
Also, the model's internal features read via the sparse autoencoders predict designability (expression, solubility, etc.) works more accurately than pLDDT
2
12
83
5,003
Daniel V Brown retweeted

Jun 9
No scaling laws for single-cell foundation models: when bigger atlases stop teaching the model anything
In language and vision, the recipe has been simple: more data, bigger models, better performance. Single-cell biology borrowed that playbook. Foundation models for transcriptomics jumped from 1 million cells to atlases of over 100 million, on the assumption that scale would unlock the same gains. Alan DenAdel and coauthors put that assumption to the test, and the result is sobering.
Working from a 22.2-million-cell corpus, they pretrained 400 models across five architectures (from PCA and a variational autoencoder up to the Geneformer transformer) and ran 6,400 evaluation experiments. They varied not just dataset size (1% to 75%) but also diversity, using cell-type re-weighting and geometric sketching to deliberately enrich rare cell types and transcriptional states.
The finding: performance saturates almost immediately. On cell-type classification, batch integration, and perturbation prediction, most models hit their ceiling at roughly 1% of the corpus, about 200,000 cells. Beyond that, adding millions more cells changed essentially nothing. More diversity didn't help. Even spiking in genome-scale Perturb-seq data, to give the models perturbed phenotypes rather than just healthy ones, failed to move the needle. Larger models did score better overall, but they too plateaued early on data.
Two points stood out. Simple baselines (PCA, logistic regression) often matched or beat the transformers. And the strongest model, SCimilarity, won not because of size but because its contrastive training objective is aligned with the downstream task. For single-cell data, what you train on and how you frame the objective matters far more than how much you collect.
This reframes a quiet but expensive habit. In drug discovery, biotech, and any pipeline leaning on cell atlases, the instinct to keep scaling pretraining corpora may be burning compute for no return. The real leverage sits elsewhere: curating high-quality, task-relevant data and matching the training objective to the actual question you're trying to answer.
Paper: DenAdel et al., journal license | doi.org/10.1038/s41592-026-0…

15
93
384
95,786
Daniel V Brown retweeted

Jun 3
It looks like there are a couple of key parameters that are critical for scaling that are not exposed in our public API.
Scaling is highly dependent on perturbing the pair representations. For the results in the paper, we set:
* Random LM dropout mask in each loop
* Random LM input mask (at the beginning)
* Random MSA subsampling (new subsample each loop)
* Random MSA column masking (modify MSA attention mask to mask out columns, changes each loop)
Working on pushing fixes for all of this and setting defaults correctly in the open source code our API.
Exact details of settings are all in the supplement.
Jun 1

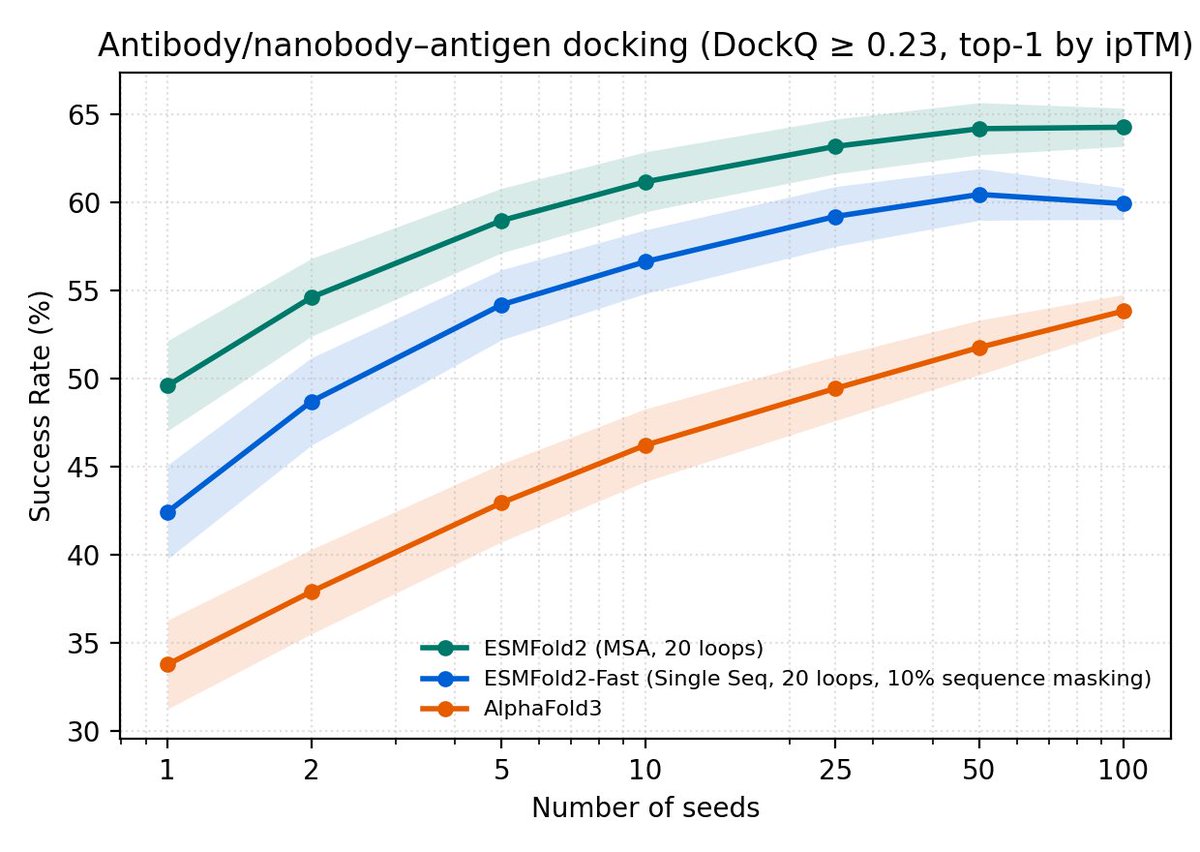
Could you share exactly what settings you're using here? I've tried to reproduce this using bound targets from this dataset (I see 106 in their metafiles), but my results are very different.
The authors actually upload AF3 scores here: github.com/NooriFatima/AF3_A…, using their data I see AF3 at 34% pass on my 106 targets.
This also appears consistent with what they report in the paper.
One caveat - I think we don't expose the 10% sequence masking flag for ESMFold2-Fast, which is important for scaling. Will push this soon.
Here's a list of all the targets I tested against: gist.github.com/rmrao/f89b69…
Will try to produce a version of this using the public API so others can also repro.
5
6
46
6,658
Daniel V Brown retweeted

How much of the human genome is essential?
Two pieces out today from our lab: 1) a method to map essential genomic intervals at gigabase scale, and 2) an argument that it's time to consider synthesizing a minimal human genome.
biorxiv.org/content/10.64898…
nature.com/articles/d41586-0…
8
47
179
40,982
Daniel V Brown retweeted

Jun 1
Binder design has come of age thanks to generative models—but how can we access the wider array of dynamic, multistate protein functions, so elegantly employed by nature?
@mihirbafna14 and I are excited to share SwitchCraft, a framework for designing such functions. (1/7)
17
143
619
84,854
Daniel V Brown retweeted

May 27
At @Biohub, our goal is to build models that accelerate scientific discovery and progress toward the cure to disease. We’re releasing all of this under MIT license allowing commercial and non-commercial use.
Read more here: biohub.ai/esm/protein/
2
32
173
30,032
Daniel V Brown retweeted

May 26
NEW preprint: Sparse autoencoders were trained on ESM-2 (650M, seq-only) and ESM-3 (1.4B, multimodal) to ask a simple question -- when two protein language models look at the same protein, do they "see" the same biology?
Answer: Yes, mostly
4
41
204
18,736
Daniel V Brown retweeted

May 20
Taken together.
Protein design has excelled at monomers, and small perfectly symmetric objects — 10-30 nm cages with defined stoichiometry.
These papers push into the mesoscale: 40-200 nm. The scale of vesicles, capsids, compartment, organelle-sized structures.
A new regime is opening up. This is where the work turns toward delivery. Programmable protein shells that enter cells with cargo — designed entirely from scratch. [14/17]
1
1
1
205
Daniel V Brown retweeted
May 20
A growing protein cage has no architect.
Each piece only responds to its local environment.
Yet somehow, thousands of pieces can come together to form a large, precise sphere.
Today in @Nature, two back-to-back papers from our team and @UWproteindesign show how de novo protein design can make this kind of self-assembly programmable.
The trick: we didn't design perfect symmetry.
We designed the rules for breaking it. [1/17]
rdcu.be/fjNgF
nature.com/articles/s41586-0…

2
31
121
9,763
Daniel V Brown retweeted

May 21
Today in @NatureBiotech we report a new suit of PE8 prime editor proteins. PE8 variants were developed from laboratory-evolved PE6 proteins using AI-guided protein redesign. This approach combines recent advances in computational protein design and directed evolution to increase prime editing efficiency, especially in transient therapeutically relevant delivery settings such as mRNA pegRNA electroporation into primary cells, eVLP delivery of prime editing RNPs, and LNP-mediated mRNA pegRNA delivery in mice.
drive.google.com/file/d/13VT…
1/11

12
92
363
82,364
Daniel V Brown retweeted

May 12
What if we could design ligands that not only bind a target, but functionally direct it toward activation or inhibition? In our #ICML2026 Spotlight, we present TD3B, a discrete diffusion framework for directional allosteric binder generation! 🦉
📜: arxiv.org/abs/2605.09810
🤗: huggingface.co/ChatterjeeLab…
6
37
221
17,210
Daniel V Brown retweeted

May 10
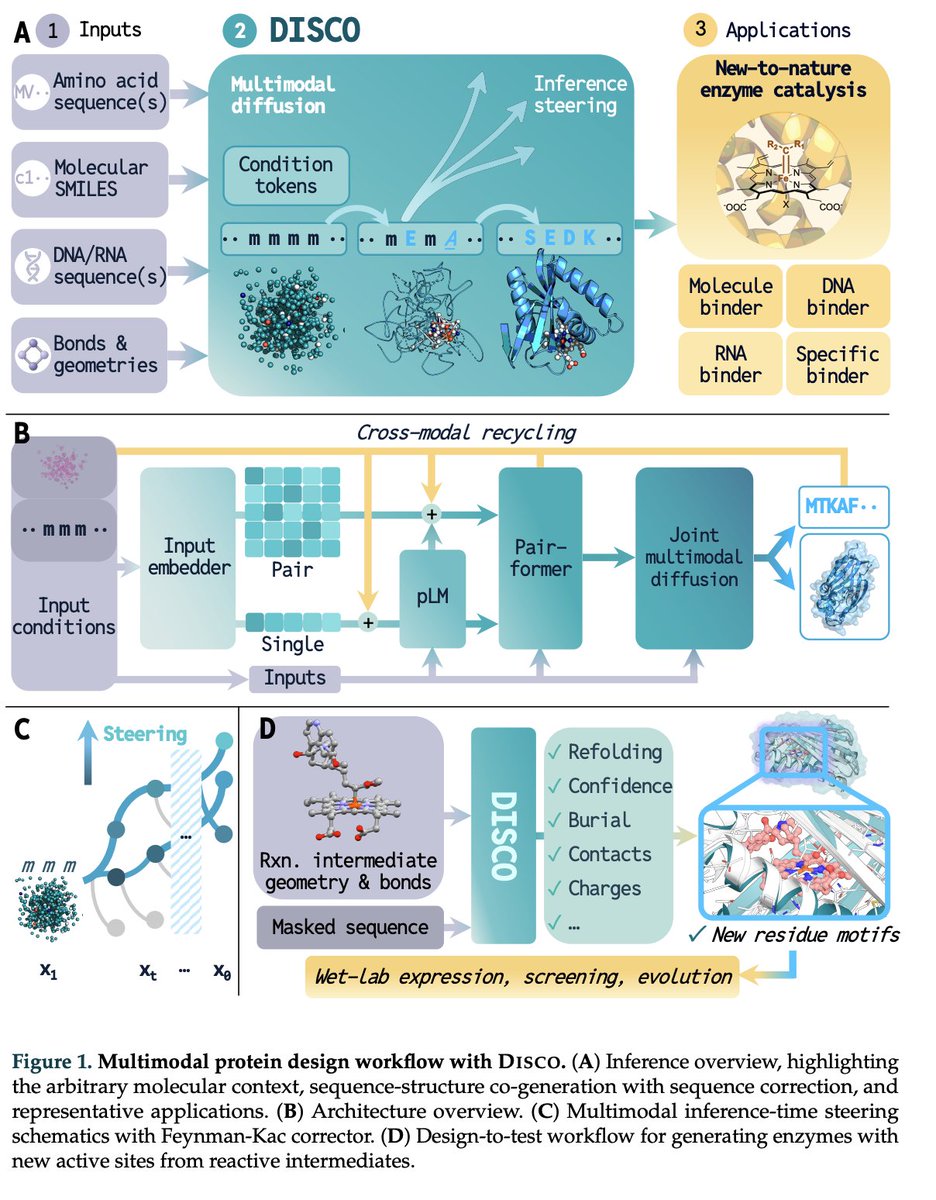
Monday Starkly Speaking with @jarridrb: a generative model designs enzymes that catalyze new-to-nature reactions — wet-lab confirmed.
"General Multimodal Protein Design Enables DNA-Encoding of Chemistry" arxiv.org/abs/2604.05181
Zoom 12pm ET / 6pm CEST: hannes-stark.com/starkly-spe…
2
23
117
16,291
Daniel V Brown retweeted

NEW: today OpenBind ‘comes out of stealth’ so to speak with their first data dump of ~900 novel protein-ligand structures - most with paired affinities
This represents a meaningful %-age increase in all of humanities P-L data in the PDB collected in the last 50 years
More👇

6
82
358
45,299
Daniel V Brown retweeted

Excited to share our paper in @NatureSMB with @antoinekoehl
Hypervariable loop profiling decodes sequence determinants of antibody stability
nature.com/articles/s41594-0…
The main question: how do antibody CDRs create enormous binding diversity without breaking folding?
4
28
120
9,177
Daniel V Brown retweeted
May 1
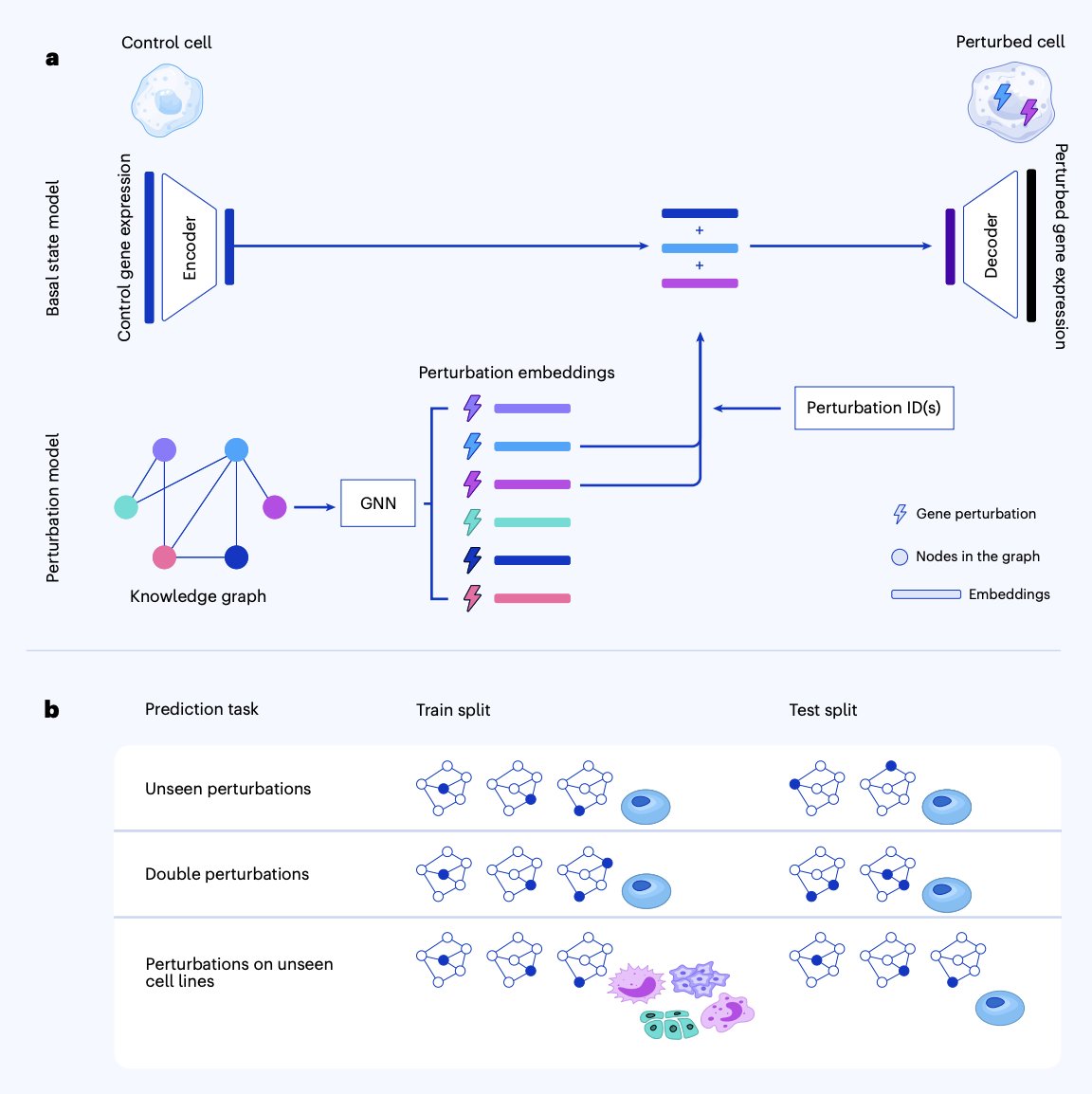
Predicting cellular responses to genetic perturbations with multiple knowledge graphs
Drug discovery is at heart a perturbation problem. You want to push a diseased cell toward a healthy state, but the space of single, double and multi-gene perturbations is enormous, and most candidates fail late at high cost. Hence the demand for ML models that predict perturbations in silico, especially out-of-distribution (new gene combinations, new cell lines), turning wet lab screens from exploratory to confirmatory. The field has been humbling: recent benchmarks showed several prominent deep models, foundation models included, beaten by trivial baselines like the training set mean of perturbation responses.
Wenkel and coauthors present TxPert, a latent-transfer framework that leans into biological priors. Two modules: a basal state encoder mapping a batch-matched control cell into a latent embedding, and a graph neural network (GAT and Exphormer variants) that learns perturbation embeddings from gene-gene interaction graphs. The two latents are summed and decoded into the predicted perturbed profile.
The key choice is combining complementary knowledge graphs: STRING and GO from curated databases, plus PxMap and TxMap from genome-scale phenomics imaging and Perturb-seq screens. Using all four consistently beat any single graph, and progressively rewiring STRING edges degraded performance monotonically, confirming the graph structure carries real signal.
Across K562, RPE1, HepG2 and Jurkat, for unseen single perturbations TxPert approaches split-half experimental reproducibility, the ceiling set by biological replicates. For double perturbations and held-out cell lines, it improves Pearson Δ by 8 to 25% over GEARS and scLAMBDA, beating the additive baseline prior deep models often failed to surpass.
For applied R&D in drug discovery and target ID, this starts to make in silico perturbation screening operationally useful. Combining curated databases with screening-derived graph priors is a reusable pattern for biotech pipelines where combinatorial screening is unaffordable.
Paper: Wenkel et al., Nature Biotechnology (2026) — CC BY-NC-ND 4.0 | doi.org/10.1038/s41587-026-0…
2
14
70
4,270
Daniel V Brown retweeted

Apr 30
Look forward to the response from the OG authors. But if this holds, it's yet another case of missing critical baselines. Also great to see folks writing formal rebuttals as preprints. This is good for science.

We benchmarked Arc Institute’s “MULTI-evolve” and found evidence that it learns a classical additive model, not epistasis.
Preprint: biorxiv.org/content/10.64898…
Blog: dewitt-lab.github.io/posts/2…
7
17
168
19,262
Daniel V Brown retweeted
Apr 30
6/ Designed proteins are no longer just static structures.
They can be cages, fibers, lattices, binders, sensors, receptor modulators, delivery handles, degradation tags, and signaling components.
Proteins are becoming a programmable medium for biology.
1
1
5
729
Apr 30
This was a really quick analysis. Hope to see more of it. Blog format really gets to the point
We benchmarked Arc Institute’s “MULTI-evolve” and found evidence that it learns a classical additive model, not epistasis.
Preprint: biorxiv.org/content/10.64898…
Blog: dewitt-lab.github.io/posts/2…
82
Daniel V Brown retweeted

Apr 9
Judging by my tl there is a growing gap in understanding of AI capability.
The first issue I think is around recency and tier of use. I think a lot of people tried the free tier of ChatGPT somewhere last year and allowed it to inform their views on AI a little too much. This is a group of reactions laughing at various quirks of the models, hallucinations, etc. Yes I also saw the viral videos of OpenAI's Advanced Voice mode fumbling simple queries like "should I drive or walk to the carwash". The thing is that these free and old/deprecated models don't reflect the capability in the latest round of state of the art agentic models of this year, especially OpenAI Codex and Claude Code.
But that brings me to the second issue. Even if people paid $200/month to use the state of the art models, a lot of the capabilities are relatively "peaky" in highly technical areas. Typical queries around search, writing, advice, etc. are *not* the domain that has made the most noticeable and dramatic strides in capability. Partly, this is due to the technical details of reinforcement learning and its use of verifiable rewards. But partly, it's also because these use cases are not sufficiently prioritized by the companies in their hillclimbing because they don't lead to as much $$$ value. The goldmines are elsewhere, and the focus comes along.
So that brings me to the second group of people, who *both* 1) pay for and use the state of the art frontier agentic models (OpenAI Codex / Claude Code) and 2) do so professionally in technical domains like programming, math and research. This group of people is subject to the highest amount of "AI Psychosis" because the recent improvements in these domains as of this year have been nothing short of staggering. When you hand a computer terminal to one of these models, you can now watch them melt programming problems that you'd normally expect to take days/weeks of work. It's this second group of people that assigns a much greater gravity to the capabilities, their slope, and various cyber-related repercussions.
TLDR the people in these two groups are speaking past each other. It really is simultaneously the case that OpenAI's free and I think slightly orphaned (?) "Advanced Voice Mode" will fumble the dumbest questions in your Instagram's reels and *at the same time*, OpenAI's highest-tier and paid Codex model will go off for 1 hour to coherently restructure an entire code base, or find and exploit vulnerabilities in computer systems. This part really works and has made dramatic strides because 2 properties: 1) these domains offer explicit reward functions that are verifiable meaning they are easily amenable to reinforcement learning training (e.g. unit tests passed yes or no, in contrast to writing, which is much harder to explicitly judge), but also 2) they are a lot more valuable in b2b settings, meaning that the biggest fraction of the team is focused on improving them. So here we are.
Apr 9

The degree to which you are awed by AI is perfectly correlated with how much you use AI to code.
1,197
2,527
20,877
4,491,978