A Semi-supervised Molecular Learning Framework for Activity Cliff Estimation
1. This paper introduces SemiMol, a novel semi-supervised learning (SSL) framework designed to enhance molecular property predictions in the presence of activity cliffs, a challenging scenario where structurally similar molecules exhibit vastly different properties. The method leverages unannotated data to improve model performance in low-data situations.
2. SemiMol employs an instructor model to evaluate the accuracy and trustworthiness of pseudo-labels generated from unannotated data. This addresses a critical issue in SSL where pseudo-labels can be unreliable due to differences between labeled and unlabeled data distributions.
3. The framework incorporates a self-adaptive curriculum learning algorithm, which progressively moves the target model towards harder samples at a controllable pace. This approach prevents the accumulation of errors from unreliable pseudo-labels and ensures robust training.
4. Extensive experiments on 30 activity cliff datasets demonstrate that SemiMol significantly outperforms state-of-the-art pretraining and SSL methods, achieving an average improvement of 26.53% in RMSE. This highlights its effectiveness in capturing chemical and biological information for accurate activity cliff estimation.
5. The study also investigates the limitations of self-supervised graph pretraining in activity cliff estimation, finding that pretraining benefits are often negligible or even negative. This suggests that SSL methods like SemiMol may be more effective in such scenarios.
📜Paper: arxiv.org/abs/2601.04507v1
#MachineLearning #SemiSupervisedLearning #MolecularPropertyPrediction #ActivityCliffs #DrugDiscovery
8
1,109

17 Nov 2025
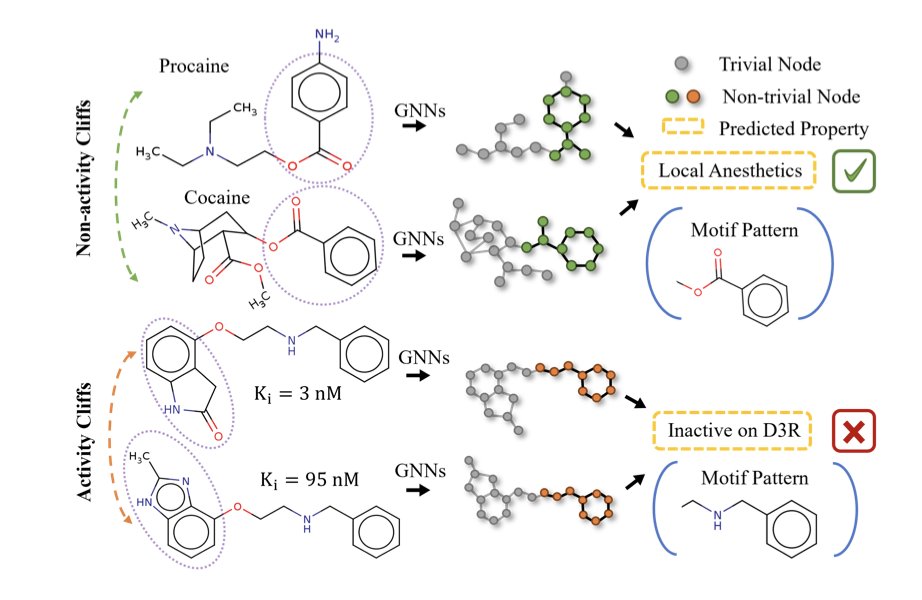
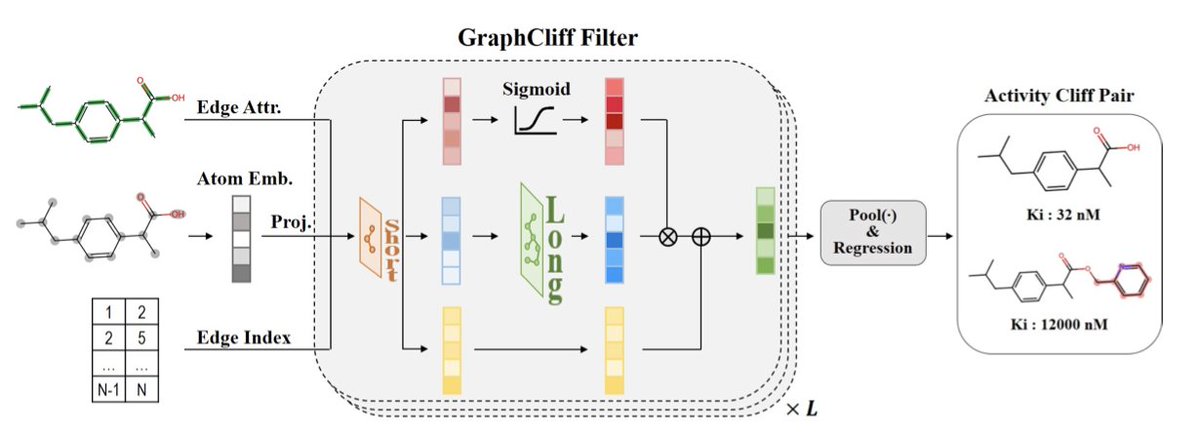
GraphCliff: Short-Long Range Gating for Subtle Differences but Critical Changes
1. A new graph neural network model, GraphCliff, has been proposed to address the challenge of activity cliffs in quantitative structure–activity relationship (QSAR) modeling. Activity cliffs refer to pairs of structurally similar compounds with large differences in biological activity, which traditional models struggle to predict accurately.
2. GraphCliff integrates short- and long-range information through a novel gating mechanism, effectively balancing local structural details with global context. This design mitigates over-smoothing, a common issue in graph neural networks, and enhances the model's ability to distinguish between structurally similar but functionally different molecules.
3. The model consistently outperformed existing graph-based models and other machine learning approaches on the MoleculeACE benchmark, demonstrating superior performance on both non-cliff and activity cliff compounds. This highlights GraphCliff's robustness in handling challenging prediction tasks.
4. Comprehensive analysis revealed that GraphCliff maintains higher sensitivity to long-range information and preserves node differentiation better than traditional GNNs. This suggests that the model effectively captures both fine-grained local variations and global dependencies within molecular graphs.
5. Qualitative analysis showed that GraphCliff's gating mechanism successfully highlights functionally relevant substructures responsible for activity cliffs, providing a more chemically meaningful representation of molecules.
📜Paper: arxiv.org/abs/2511.03170
#GraphNeuralNetworks #QSAR #ActivityCliffs #MolecularModeling #AIinChemistry
6
21
1,753
7 Aug 2025
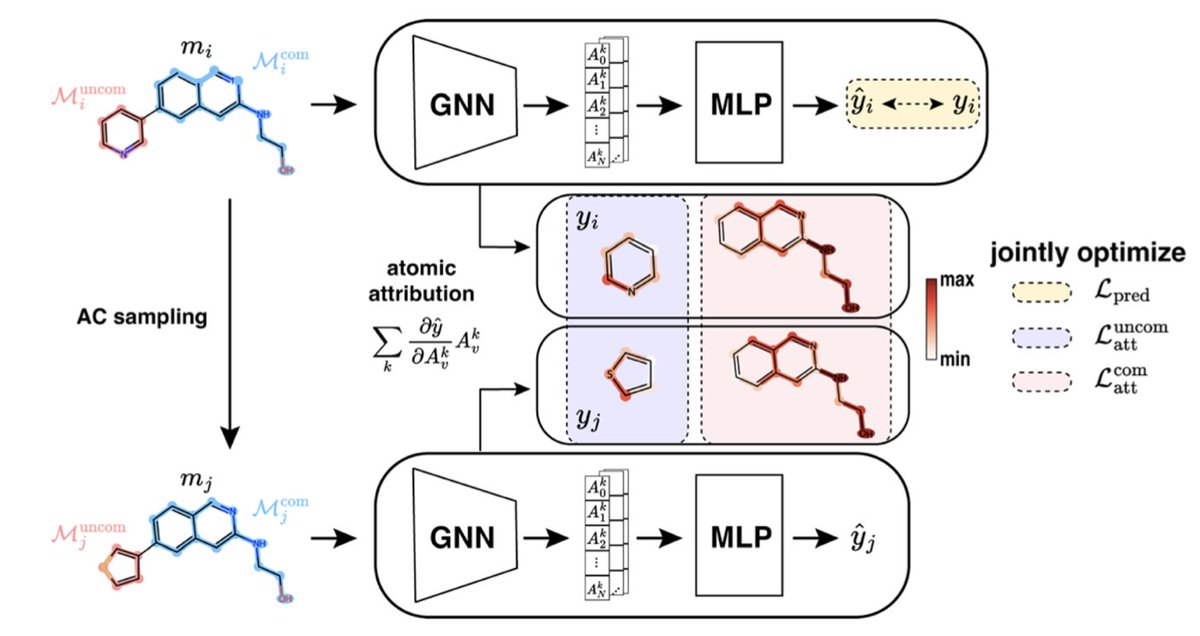
ACES-GNN: Can Graph Neural Network Learn to Explain Activity Cliffs?
1. A novel study introduces ACES-GNN, a novel framework that enhances the predictive accuracy and interpretability of Graph Neural Networks (GNNs) in molecular property prediction. This is particularly significant for drug discovery, where understanding the reasoning behind predictions is crucial.
2. The core innovation of ACES-GNN lies in integrating explanation supervision for activity cliffs (ACs) into the training process. ACs are pairs of structurally similar molecules with significant differences in potency, posing challenges for traditional models. ACES-GNN aligns model attributions with chemist-friendly interpretations, bridging the gap between prediction and explanation.
3. Validated across 30 pharmacological targets, ACES-GNN consistently improves both predictive accuracy and attribution quality for ACs compared to unsupervised GNNs. The study demonstrates a positive correlation between improved predictions and accurate explanations, highlighting the potential of explanation-guided learning in advancing interpretable AI for molecular modeling.
4. The framework incorporates an activity-cliff explanation supervision into the GNN training objective. By focusing on the uncommon substructures that explain the potency differences, ACES-GNN generates more intuitive and accurate explanations, which in turn enhances the model's ability to generalize.
5. The study also explores the impact of dataset characteristics and GNN backbones on the training scheme. It finds that explanation supervision is particularly effective in datasets with fewer analogue series, making it valuable for chemical optimization tasks.
6. The results highlight that gradient-based attribution methods can effectively integrate human prior knowledge into GNN models. Even without precise physics-based explanations, heuristic explanations for ACs contribute to better model generalization.
7. The study concludes that ACES-GNN represents a promising direction for improving both the predictivity and interpretability of GNN models in QSAR applications, with the potential to accelerate lead optimization in drug discovery.
💻Code: github.com/Liu-group/XACs
📜Paper: pubs.rsc.org/en/content/arti…
#GraphNeuralNetworks #MolecularModeling #DrugDiscovery #InterpretableAI #ActivityCliffs #QSAR
1
9
1,377
22 Apr 2025
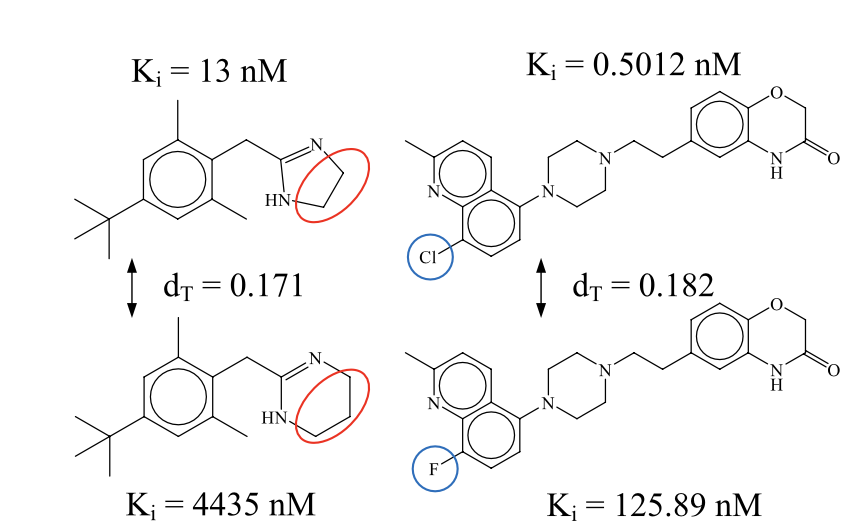
Activity cliff-aware reinforcement learning for de novo drug design
1. ACARL introduces a reinforcement learning (RL) framework that explicitly models activity cliffs—regions where small structural changes cause large shifts in bioactivity—addressing a major challenge in de novo drug design.
2. The key innovation lies in the Activity Cliff Index (ACI), which quantifies structure-activity relationship (SAR) discontinuities by comparing molecular similarity and biological activity differences, enabling systematic detection of activity cliffs.
3. ACARL integrates this ACI into RL through a contrastive loss function that dynamically prioritizes learning from activity cliff compounds, focusing optimization on high-impact SAR regions and improving molecular generation.
4. In experiments across three pharmacologically important targets (5HT1B, 5HT2B, ACM2), ACARL outperforms state-of-the-art baselines like Reinvent, JT-VAE, GCPN, MARS, and GFlowNet in generating molecules with superior docking scores and comparable diversity.
5. Unlike conventional models that treat activity cliffs as outliers, ACARL augments these critical compounds within the RL process, guiding the generative model to better capture SAR complexities and produce potent, selective candidates.
6. The framework uses a transformer-based language model pretrained on ChEMBL SMILES sequences and fine-tuned via RL, ensuring chemical validity while targeting molecules with high binding affinity.
7. Ablation studies confirm that both the contrastive loss and specific augmentation of activity cliff compounds significantly contribute to ACARL’s performance, outperforming variants without these features.
8. ACARL remains effective in multi-objective optimization scenarios, combining docking, QED (drug-likeness), and SA (synthetic accessibility) metrics, demonstrating its flexibility to adapt to real-world drug design constraints.
9. The method captures SAR discontinuities more effectively than other RL-based molecular generators, bridging a key gap in AI-driven drug design by integrating medicinal chemistry insights directly into model training.
10. While reliant on docking scores for evaluation, ACARL sets a new direction for enhancing machine learning models with pharmacological domain knowledge, offering a practical path toward discovering potent and diverse drug candidates.
💻Code: github.com/HXYfighter/ACARL
📜Paper: jcheminf.biomedcentral.com/a…
#DrugDesign #ReinforcementLearning #ActivityCliffs #DeNovoDesign #ComputationalChemistry #AI4Science #SAR #MolecularGeneration

6
25
1,761
22 Apr 2025
Activity cliff-aware reinforcement learning for de novo drug design
1. ACARL introduces a reinforcement learning (RL) framework that explicitly models activity cliffs—regions where small structural changes cause large shifts in bioactivity—addressing a major challenge in de novo drug design.
2. The key innovation lies in the Activity Cliff Index (ACI), which quantifies structure-activity relationship (SAR) discontinuities by comparing molecular similarity and biological activity differences, enabling systematic detection of activity cliffs.
3. ACARL integrates this ACI into RL through a contrastive loss function that dynamically prioritizes learning from activity cliff compounds, focusing optimization on high-impact SAR regions and improving molecular generation.
4. In experiments across three pharmacologically important targets (5HT1B, 5HT2B, ACM2), ACARL outperforms state-of-the-art baselines like Reinvent, JT-VAE, GCPN, MARS, and GFlowNet in generating molecules with superior docking scores and comparable diversity.
5. Unlike conventional models that treat activity cliffs as outliers, ACARL augments these critical compounds within the RL process, guiding the generative model to better capture SAR complexities and produce potent, selective candidates.
6. The framework uses a transformer-based language model pretrained on ChEMBL SMILES sequences and fine-tuned via RL, ensuring chemical validity while targeting molecules with high binding affinity.
7. Ablation studies confirm that both the contrastive loss and specific augmentation of activity cliff compounds significantly contribute to ACARL’s performance, outperforming variants without these features.
8. ACARL remains effective in multi-objective optimization scenarios, combining docking, QED (drug-likeness), and SA (synthetic accessibility) metrics, demonstrating its flexibility to adapt to real-world drug design constraints.
9. The method captures SAR discontinuities more effectively than other RL-based molecular generators, bridging a key gap in AI-driven drug design by integrating medicinal chemistry insights directly into model training.
10. While reliant on docking scores for evaluation, ACARL sets a new direction for enhancing machine learning models with pharmacological domain knowledge, offering a practical path toward discovering potent and diverse drug candidates.
💻Code: github.com/HXYfighter/ACARL
📜Paper: jcheminf.biomedcentral.com/a…
#DrugDesign #ReinforcementLearning #ActivityCliffs #DeNovoDesign #ComputationalChemistry #AI4Science #SAR #MolecularGeneration
4
32
1,917
16 Jan 2025
Multi-channel learning for integrating structural hierarchies into context-dependent molecular representation
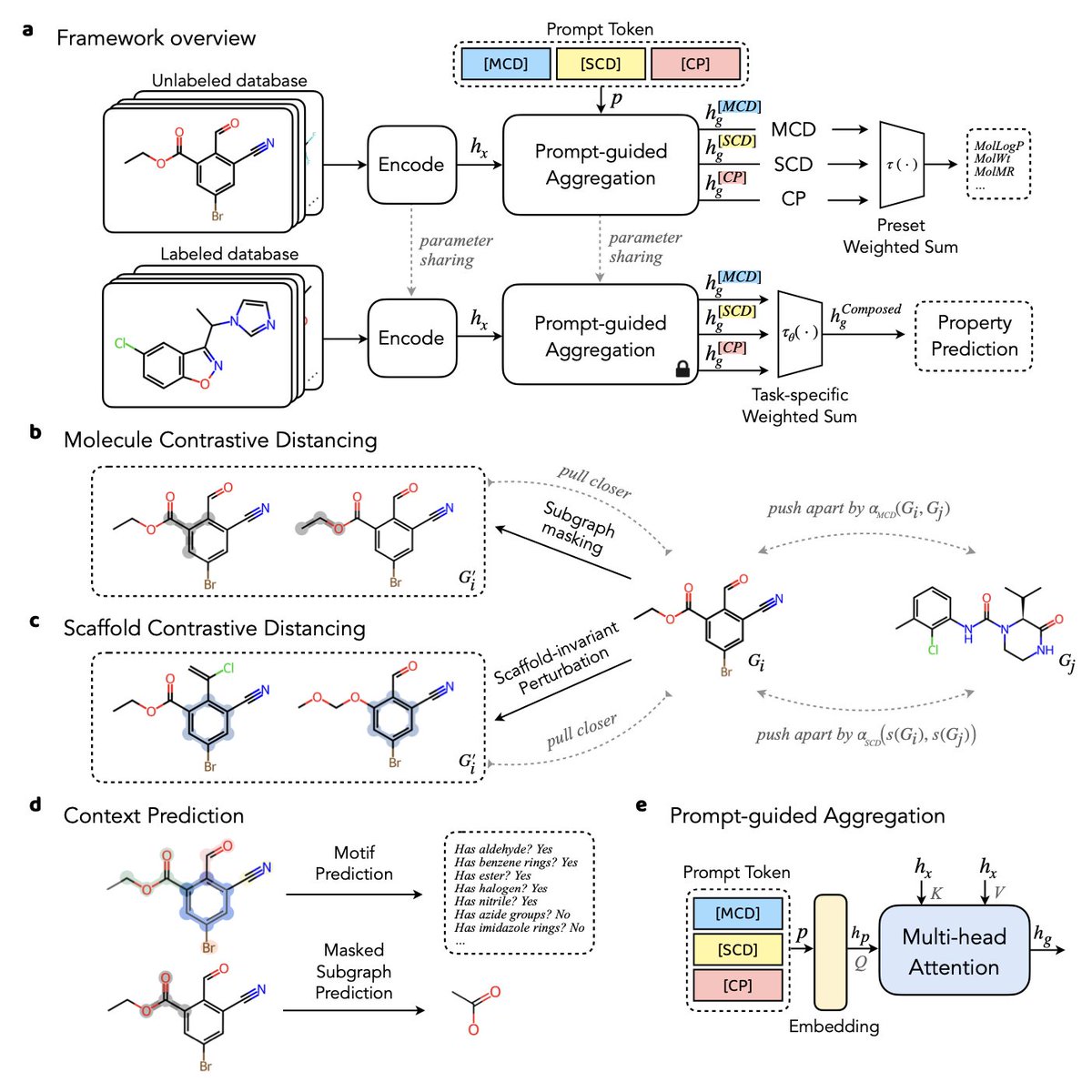
1. The paper introduces a multi-channel learning framework aimed at improving molecular representation by integrating structural hierarchies, crucial for drug discovery and molecular property prediction.
2. The framework addresses challenges like data scarcity and the complex structure-property relationships by utilizing self-supervised learning (SSL) and combining predictive tasks across multiple channels to capture both global and local structural patterns.
3. A key innovation is the prompt-guided multi-channel learning, where each channel learns a distinct self-supervised task, focusing on different aspects of the molecule: molecule distancing, scaffold distancing, and context prediction.
4. The model tackles the challenge of "activity cliffs" – situations where small structural changes significantly affect biological activity – by preserving subtle structural relationships that other methods often overlook.
5. The adaptive margin contrastive learning strategy improves the robustness of molecular representations, by calculating a dynamic margin for molecule triplets based on their structural similarity, ensuring a more nuanced representation.
6. By leveraging scaffold-invariant perturbation, the framework enhances the model's ability to learn scaffold-based similarities and push molecules with different scaffolds apart, preserving important molecular features.
7. Experimental results show that this method surpasses existing representation learning methods in performance on several molecular property prediction tasks, including BBBP, Clintox, and BACE, with notable improvements in handling activity cliffs.
8. This work demonstrates that the proposed multi-channel approach not only improves prediction accuracy but also helps in identifying key chemical patterns in molecular structure-property relationships.
9. The authors also highlight the importance of the framework's adaptability to various downstream tasks, where prompt selection helps the model focus on the most relevant information for specific applications.
💻Code: github.com/yuewan2/MolMCL
📜Paper: arxiv.org/abs/2311.02798
#MolecularLearning #DrugDiscovery #ActivityCliffs #MachineLearning #Chemoinformatics #SelfSupervisedLearning
2
13
966

14 Jan 2023
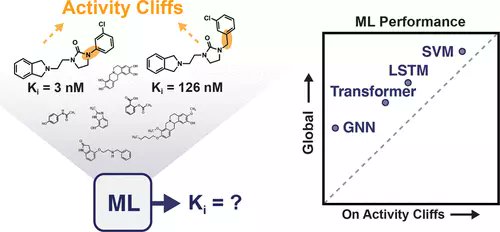
Exposing the Limitations of Molecular #MachineLearning with #activitycliffs #AI #drugdesign
pubs.acs.org/doi/10.1021/acs…
@DerekvTilborg @korney34 @fra_grisoni @molecularML
#vol62 #issue23 #JCIM #MachineLearning #DeepLearning
11
42
4,621

4 Jan 2023
Very proud to see our paper on #activitycliffs in the great review of @wpwalters! 🤩
If you missed our paper, here it is again: pubs.acs.org/doi/10.1021/acs…
w/ @DerekvTilborg, @korney34 & @molecularML
3 Jan 2023

It's back!
AI in Drug Discovery 2022 - A Highly Opinionated Literature Review
practicalcheminformatics.blo…
4
22
3,091
27 Jan 2022
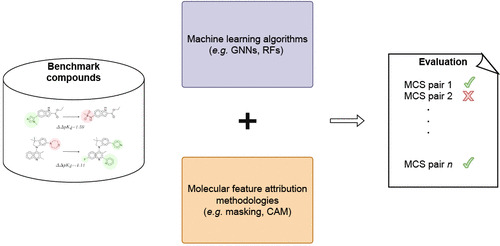
Benchmarking Molecular Feature Attribution Methods with #ActivityCliffs #moleculardesign #ArtificialIntelligence
pubs.acs.org/doi/10.1021/acs…
@josejimlun @Mih4S90 @NilsWeskamp
#current_issue #MachineLearning
4
13

16 Dec 2021
"Learning functional group chemistry from molecular images leads to accurate prediction of activity cliffs" Available on @AILSCI!
🔗Find out more at: doi.org/10.1016/j.ailsci.202…
#ActivityCliffs #prediction #AI #DeepLearning #compchem #chemtwitter #openaccess
3
3

27 May 2019
#ActivityCliffs are of great interest & useful for testing models but it will the ability to reliably predict relative affinity rather than relative affinity for affinity for activity cliffs that will change how #CompChem will impact optimization in #DrugDesign #alchemy2019
1
1

12 Jun 2018
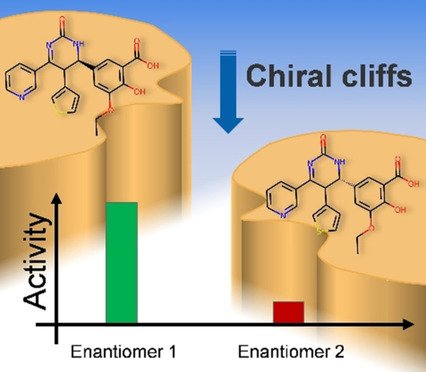
Schneider, Ertl et al. @Novartis report their new 2D descriptors for building predictive models, via machine learning, to identify activity cliffs in pairs of #enantiomers #activitycliffs #drugdiscovery #machinelearning #compchem doi.wiley.com/10.1002/cmdc.2…
3
6
6 Nov 2017
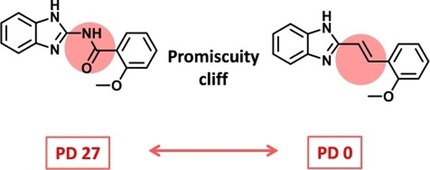
Distinguishing bad from good compound promiscuity @LIMES_Bonn #ActivityCliffs #MedChem doi.wiley.com/10.1002/cmdc.2…
2
7