Decoding Synonymous Codon Selection With a Transformer Model
1 CaNAT is a non-autoregressive Transformer that predicts an entire codon sequence directly from an amino-acid sequence, with a design choice that prioritizes rare-codon learning via batch-wise weighted cross-entropy (rare codons contribute more to gradients instead of being drowned out by frequency bias).
2 Trained at scale on >3 million coding sequences from 620 species (taxonomically broad, cluster-split to avoid homology leakage), CaNAT aims to recover native synonymous choices rather than only “optimize” codons for expression, and it does so without providing organism labels during training.
3 A key practical output is a codon-level confidence score (softmax probability per position). The study shows confidence correlates strongly with accuracy, and introduces a degeneracy-aware threshold T(k, α) to compare confidence fairly across amino acids with different numbers of synonymous codons.
4 On the full multi-species test set, CaNAT reaches ~53% exact codon accuracy, beating statistical baselines such as “always pick the most frequent codon” (~48%), frequency sampling (~39%), and uniform random (~33%). High-confidence filtering further boosts accuracy.
5 Compared to CodonTransformer (an organism-fine-tuned model), CaNAT is especially strong on rare codons (RSCU < 0.7): across shared benchmark species, CaNAT generally improves rare-codon prediction (notably in human and mouse), suggesting the balanced-loss strategy helps capture context-dependent rare-codon placement.
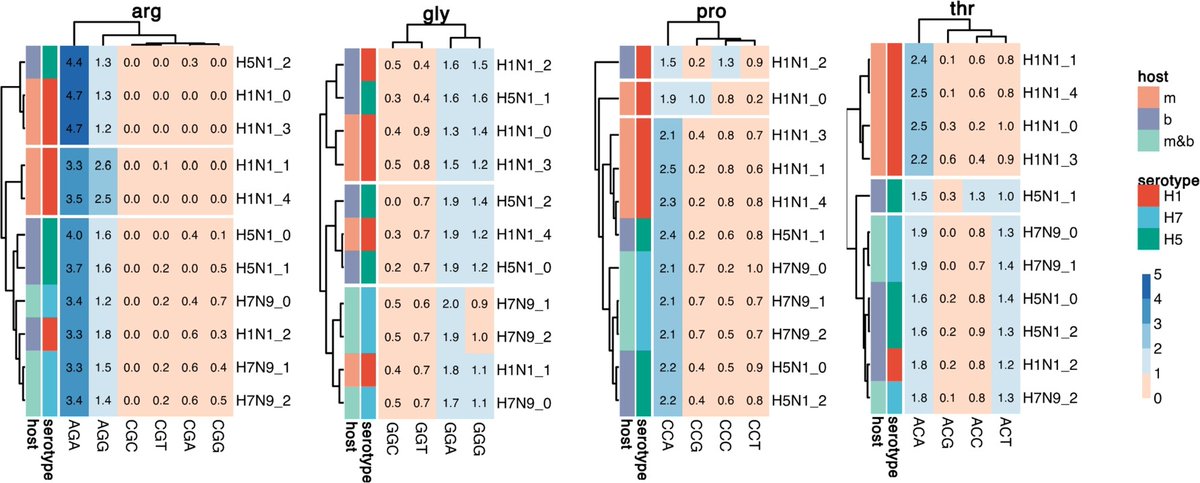
6 Even without species labels, CaNAT’s predictions recapitulate organism-specific synonymous codon distributions: for E. coli, H. sapiens, and an extremophile (Streptococcus thermophilus), predicted vs observed codon-usage profiles show near-perfect rank agreement across amino acids (median Spearman close to 1), including preservation of rare codons.
7 Internal representations encode species identity at positional resolution: embeddings from the decoder separate species via LDA, implying that local sequence context contains enough signal for the model to infer organism-specific codon “signatures” throughout a gene.
8 The model captures biophysical constraints beyond GC-content: adding RNA stability (ViennaRNA) to a regression explaining prediction accuracy increases explained variance from R²=0.148 (GC only) to R²=0.191 (GC stability), indicating that codon-choice predictability relates to structured RNA features.
9 Attention analysis reveals multi-scale context for codon choice: (i) tight near-diagonal heads consistent with dicodon/codon-pair effects, (ii) broader local windows spanning several codons, and (iii) long-range diagonals with offsets roughly −70 to 60, suggesting distant regions contribute to local synonymous decisions; attention is often biased slightly downstream.
10 CaNAT predictions connect to functional constraint: on three deep synonymous-scanning datasets in E. coli (ddlA folding efficiency, RNase III function, TEM-1 β-lactamase fitness), accuracy is highest at “wild-type only tolerated” positions, which are enriched for rare codons; at partially tolerant sites, CaNAT often predicts an alternative tolerated codon rather than exactly the wild type, consistent with learning constraint strength rather than memorizing sequences.
💻Code:
github.com/Andre-lab/CaNAT/
📜Paper:
biorxiv.org/content/10.64898…
#ComputationalBiology #CodonUsage #SynonymousMutations #Transformers #DeepLearning #Translation #RNAstructure #ProteinFolding #Bioinformatics #Genomics