
#Deucalion May newsletter is now available:
- Fsas Technologies’ visit to #Deucalion
- The impact of hosting the #EasyBuild User Meeting
- Participation in the EPICURE #Hackathon and Jornadas FCCN
- Deucalion's role in the #EuroHPC Federation Platform
➡️ shorturl.at/7tEqi

2
21


Apr 23
=>
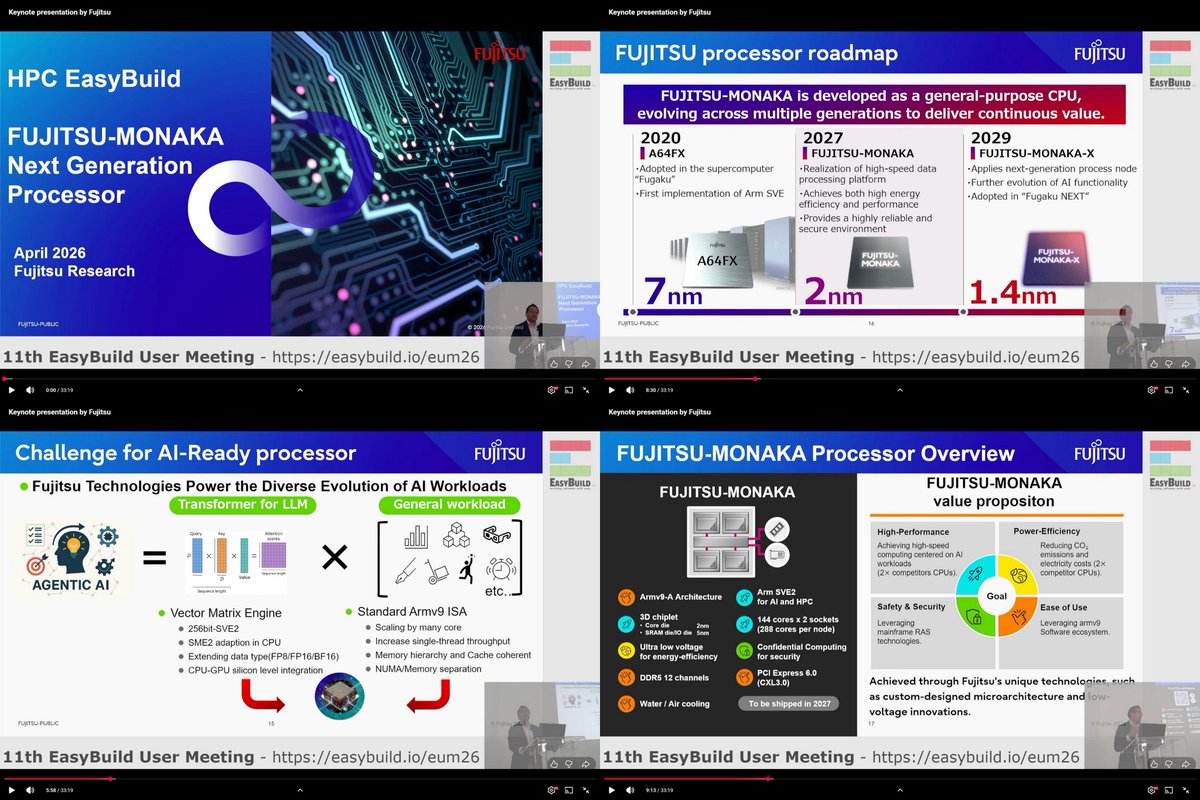
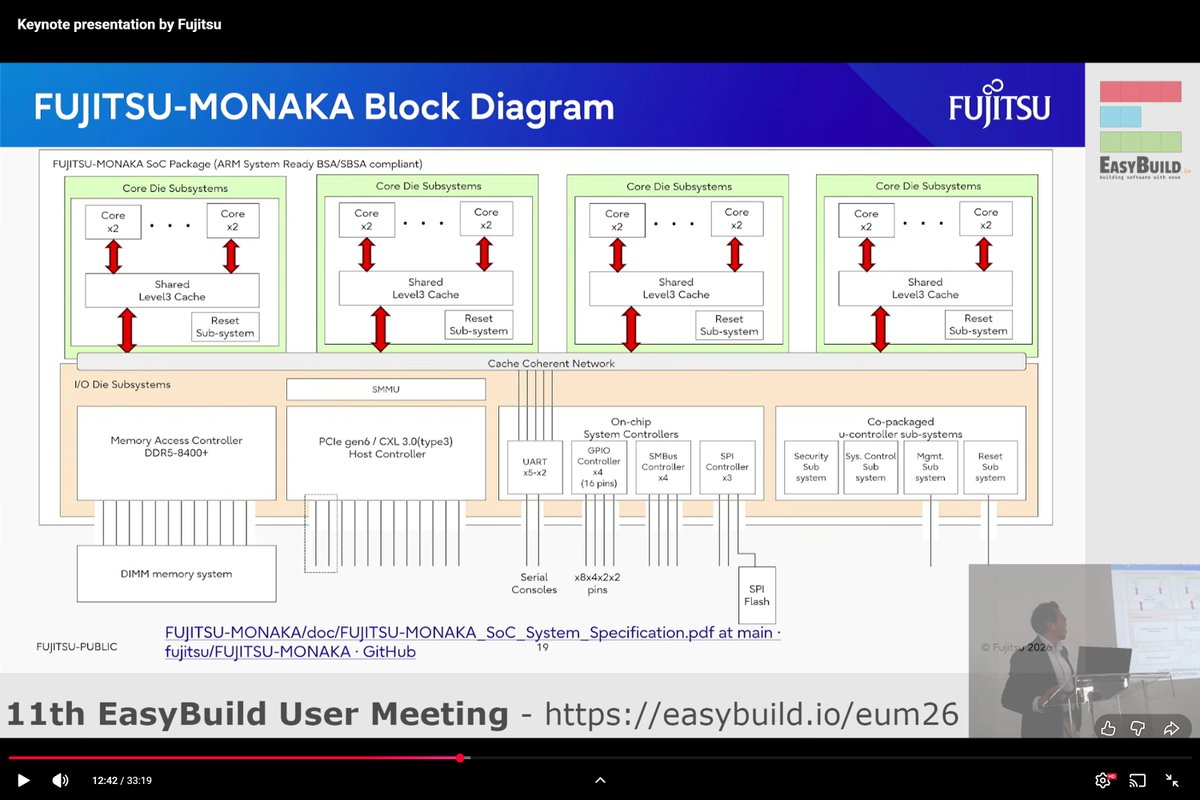
"FUJITSU-MONAKA Next Generation Processor", Satoshi NAKAJIMA, Fujitsu, Keynote, EasyBuild User Meeting, Apr 23, 2026 youtube.com/watch?v=QyF3Em6w…
BRCM, Feb 26 x.com/ogawa_tter/status/2027…
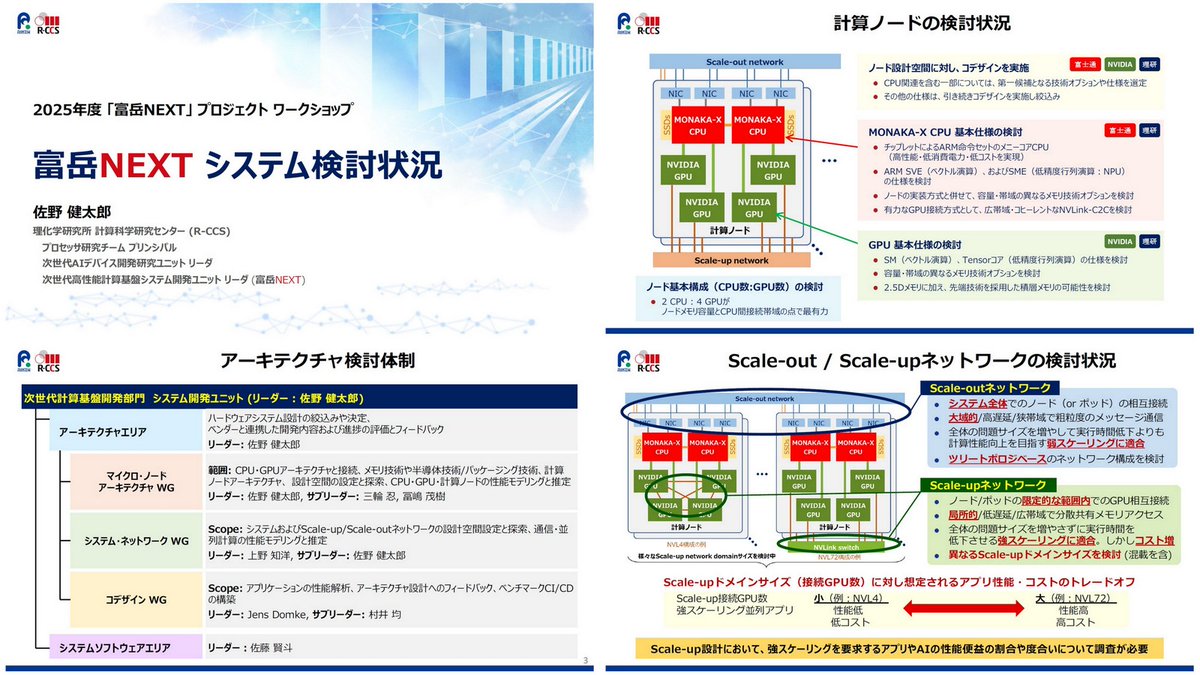
2025年度「富岳NEXT (FugakuNEXT)」プロジェクト WS、3月6日 x.com/ogawa_tter/status/2034…
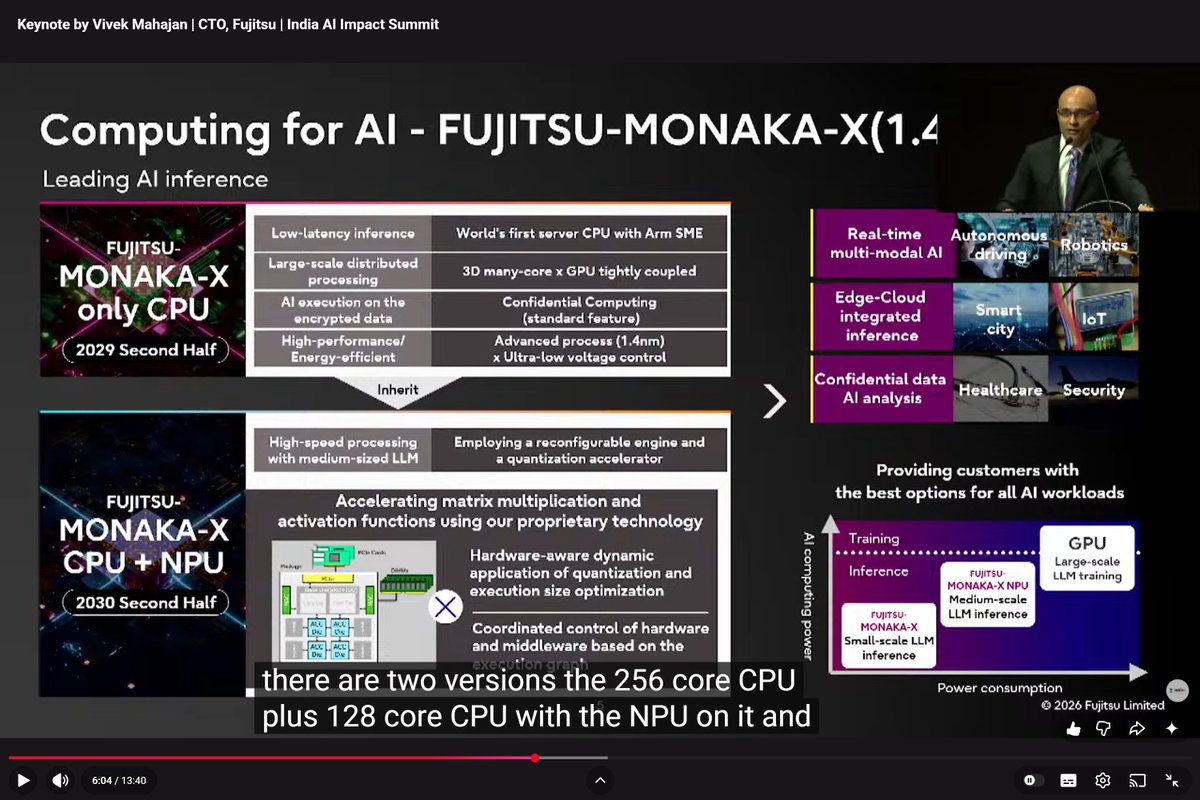
MONAKA-X, Feb 20

Mar 19

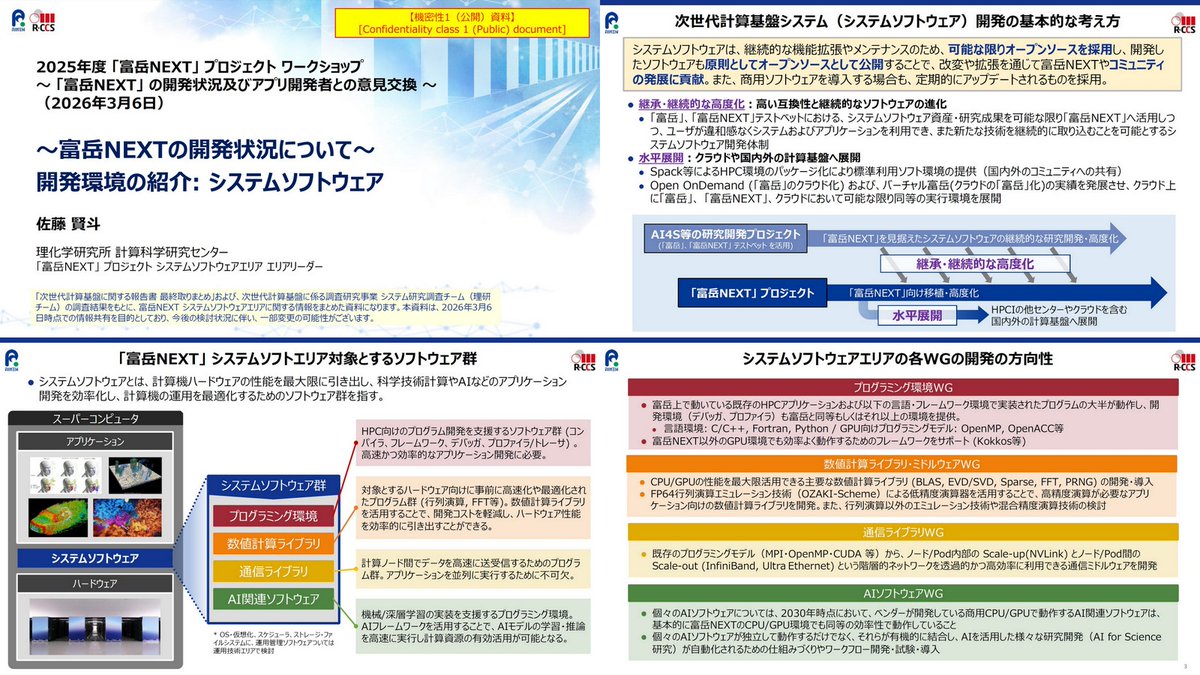
2025年度「富岳NEXT」プロジェクト WS、2026年3月6日
全体説明 r-ccs.riken.jp/events/202603…
システム検討 r-ccs.riken.jp/events/202603…
開発環境
r-ccs.riken.jp/events/202603…
r-ccs.riken.jp/events/202603…
R7年度補正 (成立) x.com/ogawa_tter/status/1995…
373億円
<= MONAKA-X, Fujitsu, Feb 20


1
3
595

EasyBuild and Spack fetch the source and build locally. Do you mean Python packages that are not open-source?
2
2
33

7 Nov 2025
We are excited to announce the Call for Participation for the Package Managers devroom at @fosdem 2026, taking place on Saturday, 31st January 2026 at the Université libre de Bruxelles, Belgium.
Submission deadline: 1st December 2025
The Free and Open Source community has built dozens of package managers across operating systems (Debian's APT, Fedora's DNF, Arch's pacman, Nix, Guix), programming language ecosystems (PyPI/pip/uv, npm, Cargo, RubyGems,
Conda, Pixi), domain-specific tools (Spack, EasyBuild for HPC), and universal formats (Flatpak, Snap, AppImage). Each has made unique trade-offs and innovations.
This devroom provides a gathering place for maintainers, researchers, and users of package managers to discuss lessons learned, new developments, and common infrastructure challenges. It's a neutral forum where different
communities can meet, exchange ideas, and foster collaboration on universal issues.
SUBMISSION GUIDELINES
Submit your proposal through the FOSDEM pretalx system:
pretalx.fosdem.org/fosdem-20…
1
2
105

6 Nov 2025
👶 Hey kiddo! Wanna build a super fun app, like a game where puppies chase rainbows? 🐶🌈
But... scary computer stuff? No way!
**Coding Kickstarter** is your magic robot friend! 🚀
Tell it your big dream (easy words only).
Robot says: "Yay! Here's 5 happy steps. Copy! Paste! Build! No ouchies."
Like drawing with crayons, but for apps. Try now: codingkickstarter.com
What's YOUR dream app? Tell me! 😄
#KidCoder #EasyBuild

3
605

3 Oct 2025
We broke down affordable housing with solutions like EasyBuild, navigating entrepreneurship vs employment & exploring life assurance. This edition turned real-life questions into practical answers.
#NCBATwendeMbele #CustomerObsession




1
1
2
297

19 Sep 2025
Big news for real estate investors: Easy Street Capital has increased RTL loan amounts!
Now up to $5M for EasyFix (fix & flip) and EasyBuild (new construction projects). Get the funding you need to scale. Competitive rates. Fast closings.
#RealEstateInvesting #HardMoneyLoans

2
45
19 Aug 2025
NCBA EasyBuild is our one-stop solution for home construction. With up to 100% financing, a 9-month principal grace period, professional supervision, and customizable designs, we make your home ownership journey seamless.- Philip Omondi
x.com/i/spaces/1MYxNlZYQQZGw
#NCBATwendeMbele
2
2
433
1 Aug 2025
Breaking down affordable housing and EasyBuild, planning across entrepreneur versus employment journeys, and life assurance. That’s what we offered, answering how you plan when life keeps changing.
#NCBATwendeMbele #Goforit


1
1
3
375

🖤💙 THANK YOU 💙🖤
A massive thank you to our club sponsors - you’re behind our successful operation and day to day running. We would be nowhere without you!
Our thanks go to:
Sports Lighting - @SportsLEDs
Easybuild
Precision Decking
Devon Hills - @Devon_Hills
Wollens
Everyone at Stoke Gabriel & Torbay Police FC thanks you!!

4
910
1 Aug 2025
Next: Swindon!
Connect with NCBA to learn about diaspora solutions and investing back home with solutions such as EasyBuild.
Sign up here: forms.office.com/r/JT4jnXS3v…
#NCBADiasporaConnect #GoforIt

2
338

Did you know that @NCBABankKenya's Affordable Housing Prog home loan is designed for individuals earning up to Kes200K per month and it offers options such as straight purchase, construction, buy & build, and EasyBuild options, with a loan cap of Kes10.5M?
1
2
4
1,085

26 Jun 2025
.@NCBABankKenya offers an affordable home loan for individuals earning up to Ksh. 200,000 per month. It offers options such as straight purchase, construction, buy & build, and EasyBuild options, with a loan cap of Ksh. 10.5 million. hapakenya.com/2025/06/26/you…
2
1
193

19 Jun 2025
Engineering Supercomputing Platforms for Biomolecular Applications
1.This extensive benchmarking study reveals that Nvidia's GH200 “Grace Hopper” superchip delivers an order of magnitude better molecular dynamics (MD) performance than its predecessor, the V100, across all tested software — including NAMD, GROMACS, AMBER, LAMMPS, and OpenMM — while also being more energy-efficient.
2.While AMD's MI250X GPUs offer competitive raw performance at lower cost, their ROCm software ecosystem is still immature, requiring more developer effort and resulting in compatibility issues for some major MD codes like OpenMM and partial LAMMPS support.
3.The authors stress that raw FLOPS no longer reflect real-world performance. Instead, matching hardware and software to the specific needs of biomolecular methods — MD, quantum chemistry (QC), electron microscopy (EM), and coarse-grained MD — is key to maximizing efficiency and usability.
4.Single-GPU execution remains optimal for most MD workloads. Jobs run faster and more efficiently when parallelized into many independent tasks on single GPUs rather than scaling to multi-node executions, which can triple energy use per nanosecond of simulation.
5.Multi-instance GPU (MIG) capabilities in Nvidia's A100 allow multiple MD simulations to run in parallel on a single GPU without performance loss. AMBER showed strong scaling up to 28 replicas, while GROMACS suffered from CPU contention, limiting its scalability.
6.The report highlights severe performance and compatibility issues when deploying Cryo-EM pipelines (e.g., RELION) on traditional HPC systems. These include software instability, MPI configuration problems, and tight storage limits (e.g., 500 GB quota vs. average 2.5 TB dataset).
7.Quantum chemistry benchmarks using Psi4 showed GH200 CPUs excel in simple basis sets, but face instability with more complex configurations. Memory bandwidth and CPU architecture significantly influence performance in CCSD calculations.
8.Energy efficiency is a central theme: GPUs are more efficient than CPUs for MD, but excessive node scaling harms power efficiency. Even efficient hardware needs software optimized for energy-aware execution.
9.Coarse-grained MD (CGMD) remains underutilized on HPC due to limited software support. Many models rely on homegrown code that lacks portability and scalability, posing barriers to broader adoption unless integrated into major MD frameworks.
10.The authors argue that sustainable HPC must be seen as a people problem. Expertise in DevOps, better system administration, user training, and community-driven consortia (like HECBioSim) are vital to reduce downtime, improve support, and accelerate science.
11.They call for continued and expanded support of the consortium model, especially in preparing for even more complex future hardware (e.g., MI300, Intel GPUs, and Nvidia’s AI-accelerator class CPUs).
12.The report also critiques current HPC queueing and provisioning systems, which reward short, large-scale jobs over long, efficient single-node jobs, thus wasting compute and power.
13.Finally, the authors suggest making the HPC environment more user-friendly through widespread adoption of build frameworks (EasyBuild, Spack), containerization (podman-hpc), and better data transfer/storage solutions.
📜Paper: arxiv.org/abs/2506.15585v1
#HPC #MolecularDynamics #CryoEM #QuantumChemistry #AMD #Nvidia #GH200 #ROCm #EnergyEfficiency #DevOps #HECBioSim

3
4
791
15 Jun 2025
Customers explored the pros and cons of owning vs. renting, with a spotlight on our Property Finance solutions, including Affordable Housing and NCBA EasyBuild.
#NCBATwendeMbele #Goforit


1
1
540

POP Expert Brian Wylie of @fzj_jsc introducing the POP CoE at the EasyBuild User Meeting 2025
@easy_build @EESSI_hpc @MultiXscale


2
2
105

25 Feb 2025
‼️ NEW PLAN ALERT‼️
Build some ana-white.com/woodworking-pr…
#AnaWhite
#EasyBuild
#DIYPlanterBox
#WoodworkingProject
#HandmadeGarden
#SimpleBuild
#BeginnerFriendly
#PlanterBoxDIY
#GardenProjects
#WeekendWoodworking
#OutdoorPlanter

1
1
4
556

25 Feb 2025
The 2nd beta release of EasyBuild v5.0.0 (EasyBuild v5.0.0beta2) is now available!
Please consider testing this beta release...
More details, including highlighted changes, installation instructions, planned additional changes are available via docs.easybuild.io/easybuild-…
#HPC
2
77
If you actively use or contribute to EasyBuild, please participate in the 8th EasyBuild User Survey:
ugent.qualtrics.com/jfe/form…
Help us reach our goal of at least 100 participants by the end of the month!
#HPC
1
4
140