


Jun 16
A structure-guided pipeline yields peptide inhibitors that disarm fungal peptidase-driven virulence and resistance
"... our computational pipeline designed putative inhibitors based on the ipTM and LIS scoring metrics for each of the targeted cryptococcal peptidases with modifications to increase target specificity and inhibitory activity.
...
Across evaluation datasets, ipTM and LIS showed the strongest performance; however, LIS consistently achieved a slightly lower FPR at comparable TPRs for peptide–peptidase complexes, while both metrics performed equivalently for protein datasets, supporting LIS as a more reliable indicator for peptide–peptidase prediction."
biorxiv.org/content/10.64898…
6
16
1,328

Jun 16
虽然我本人不喜欢颜宁,但从科学角度评价,她的工作确实做得非常漂亮,就算去掉解析那一步,光是纯化蛋白到能上机的水平,全球至少95%的实验室都做不到。觉得结构生物学就是拍张照的人,建议自己去AlphaFold跑几个膜蛋白试试。看看pLDDT低于50是什么概念,看看PAE高得离谱是什么样子,再看看ipTM score有多惨。另外,其工作重点之一是通过实验捕获蛋白处于不同工作状态的动态构象变化,这也是AlphaFold这类静态预测工具目前无法实现的。生物学是靠大量人力和真实数据累积出来的。AI能提供方向性参考,生产出的大量假阳性结果必须要实验来验证。幻想AI取代真的很蠢

10
1
73
36,144

Jun 15
MoE-Bind: Guiding De Novo Protein Binder Generation with Sparse Experts
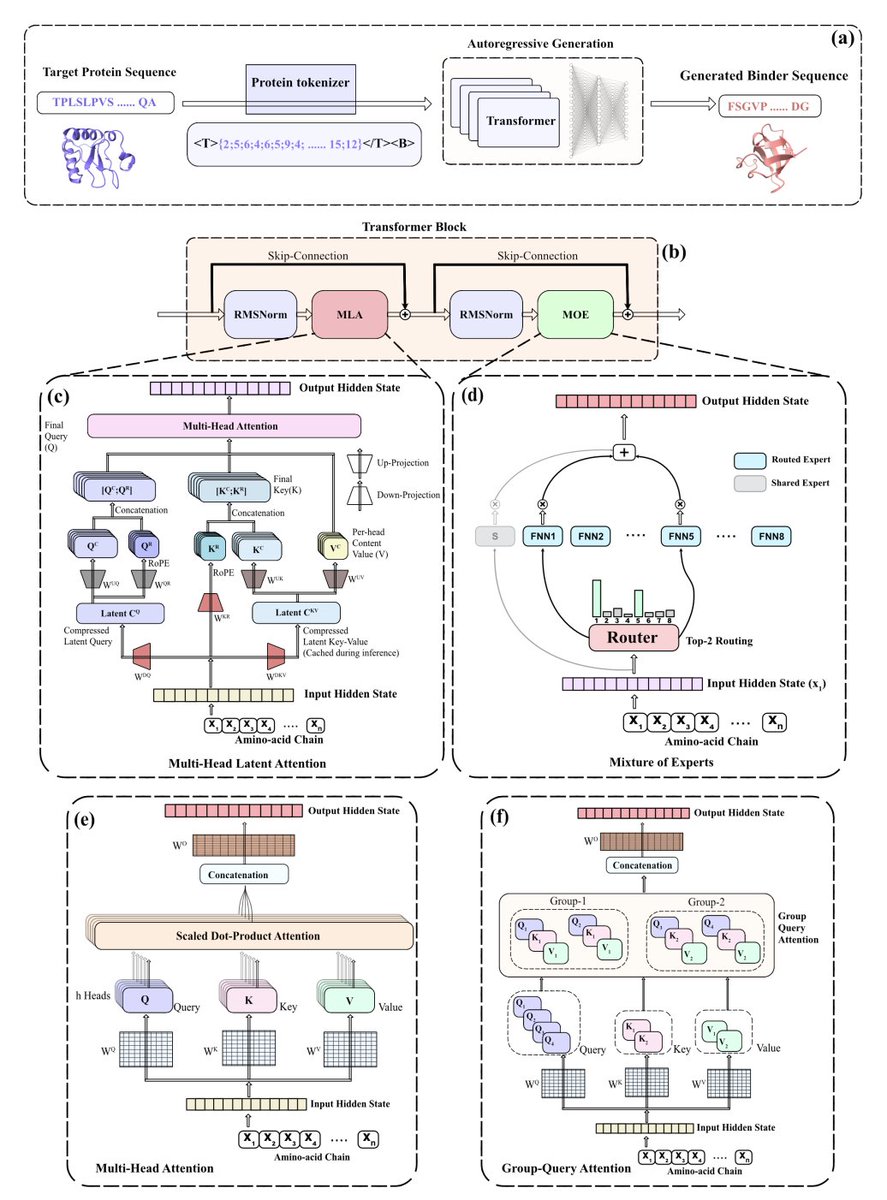
1. The paper introduces MoE-Bind, a sequence-only autoregressive protein binder generator that combines Multi-head Latent Attention (MLA) with a sparse Mixture-of-Experts (MoE) feed-forward stack, aiming to keep binder generation fast and structure-free at inference while improving quality per unit compute.
2. Key architectural idea: sparsify where most parameters live. Since transformer FFNs hold a large fraction of parameters, MoE-Bind replaces dense FFNs with top-2 routing over 8 SwiGLU experts (plus a shared always-on expert), so only ~2/8 of expert parameters activate per token while total capacity increases.
3. MLA targets the other bottleneck: KV-cache memory during autoregressive decoding with long receptor prompts. MoE-Bind compresses keys/values into a low-rank latent (rKV=64) and uses decoupled RoPE (separate positional subspace), yielding a large KV-cache reduction (reported 24× vs a GPT2-like MHA peer at the 100M tier).
4. Compute/parameter framing: the 100M-parameter MoE-Bind model has ~102.7M total params but ~38.8M active params per token, positioning it as “compute-matched” against ~38M dense baselines while often matching or exceeding ~100M dense baselines in structure-level metrics.
5. Training pipeline: pre-train on UniRef50 (character-level tokenization; 31-token vocab including delimiters/control tokens) with next-token prediction, then instruction fine-tune on high-confidence STRING v12 physical PPIs (score ≥900) after heavy redundancy reduction (MMseqs2 clustering at 40% identity, 80% coverage), ending with ~2.1M usable interaction pairs.
6. Leakage control is a major methodological emphasis. For DB5 evaluation, the authors build a strict 22-target benchmark by removing any DB5 proteins with ≥10% identity (≥80% coverage) to UniRef50 or STRING sequences, then also report a larger benchmark (78 unique targets) under a relaxed fine-tuning-only leakage filter and additional deduplication.
7. Structure-level evaluation uses structure predictors only for external assessment, not for inference-time filtering: AlphaFold2-Multimer (ColabFold) on the strict 22-target DB5 set, and Boltz-2 with MSA on the larger 78-target set. Hits are defined stringently as generated ipTM ≥ reference (native pair) ipTM for the same target.
8. Main structure-level results: on the 22-target AF2-Multimer evaluation, MoE-Bind achieves 6/22 hits (27.3%) vs MHA 3/22 and GQA 4/22; on the 78-target Boltz-2 MSA benchmark, MoE-Bind reaches 19/78 hits (24.36%), slightly higher than dense 100M baselines (GQA-100M 23.08%, MHA-100M 21.79%) and higher than compute-matched dense ~38M baselines (GQA-38M 20.51%, MHA-38M 16.67%) while activating ~38.8M params/token.
9. Sequence-level quality: MoE-Bind’s generated binders better match DB5 amino-acid composition, avoid long homopolymer runs (no 6–7 or ≥8 runs reported), show “controlled novelty” vs STRING (less mass at ~0% identity than dense baselines), and have improved predicted stability by instability index (median ~29–30, with ~2/3 below 40).
10. Interpretability contribution: routing analysis reports expert specialization at individual amino-acid and biochemical-group levels, arguing that proteins’ small, biochemically structured alphabet makes MoE routing more interpretable than typical natural-language MoE behavior, and suggesting future expert pruning/specialization guided by biochemical priors.
📜Paper: biorxiv.org/content/10.64898…
#ComputationalBiology #ProteinDesign #ProteinEngineering #ProteinLanguageModels #MixtureOfExperts #Transformers #DeepLearning #Bioinformatics #PPI #DeNovoDesign
5
32
2,371

Sure, that's good. ipSAE solves the problems with ipTM pretty well. Basically, depends on whether there are any PAE<cutoff (usually 10 Å) and depends on how many amino acids have PAE<cutoff. Would be great to add to colabfold.
1
91

Jun 14
I'll work on exposing the distogram features. That should solve most of the problems people are seeing with iptm/ipae.
1
1
62
Stop using ipTM for PPI... :-).
1
51

Jun 14
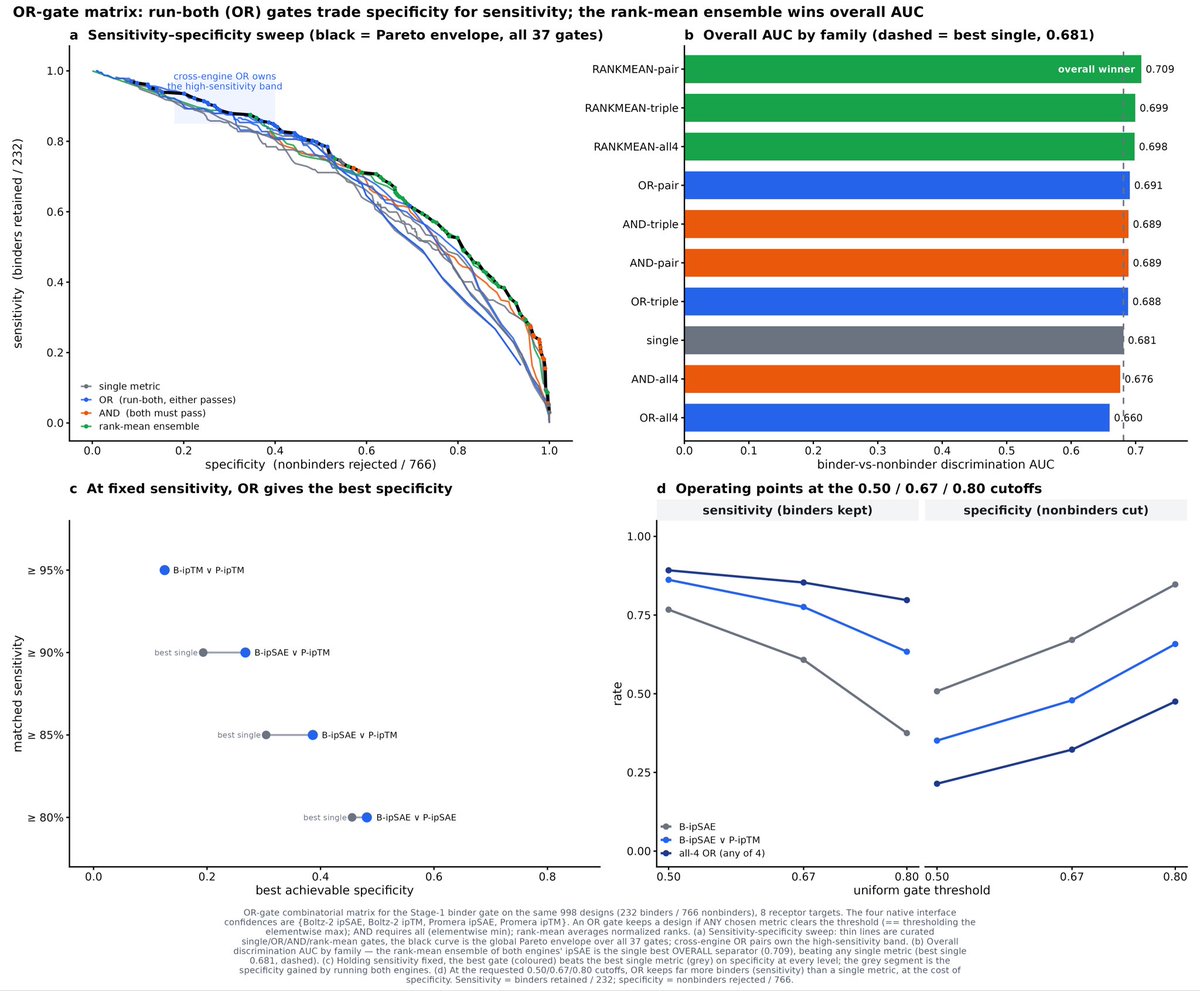
Of course, trading off specificity vs. sensitivity is key for experimental design. A sweet spot of structural metrics is able to discriminate 2/3 of binders AND eliminate 2/3 of non-binders.
ipSAE reigns supreme for discriminating non-binders while ipTM keeps false positives.
1
3
366
Jun 14
DeltaForge is able to reliably discriminate top performing from worst performing binders, boosting signal and discrimination beyond ipSAE and ipTM scores alone.

1
6
443
The real Boinc Italy - Boinc in Italiano retweeted
Jun 11
Promera (@bjing2016) is the first folding model aside from Boltz-2 that is reliably able to produce strong poses, ipTM, and ipSAE scores for LigandForge binders. How do ESMFold2-Fast, OpenFold3, Protenix-V2, Boltz-2 and Promera compare?

Jun 11

@mihirbafna14 and I are excited to introduce Promera, a co-folding and design model with
• best-in-class binder filtering
• nanobody design with in-silico success rates matching hallucination
• case studies on hantavirus epitope targeting and GPCR agonism (1/8)
6
13
70
12,766

Jun 14
I don’t get it.
HalluDesign-NA is interesting, but it feels more like a pipeline remix than a new design breakthrough.
Take HalluDesign, swap in NA-MPNN, use AF3/Protenix as the scorer, then optimize pLDDT / pTM / ipTM.
That’s not RFdiffusion3 for nucleic acids.
RFdiffusion-style models generate structures.
This is more like using AF3 as a reverse-design heuristic.
Fine idea. But without experimental aptamer validation, a pretty predicted DNA/RNA structure is still just a pretty prediction.
Jun 12

HalluDesign-NA: Extending HalluDesign for De Novo Nucleic Acid Design biorxiv.org/content/10.64898…
1
74
Luminary Myers ࣪𖤐 retweeted

Jun 14
@Kill3rvfx que racista ve los comentarios que haces “no podes ser tan mexicano Iptm” dice ¿tienes algo de malo ser mexicano? @alpamel11r


4
7
12
1,670
CUI LUN🇫🇷🇨🇳 retweeted

Jun 9
ipTM and ipSAE don't predict binding affinity.
A-alpha Bio measured 7M interactions: almost no correlation.
Dug into this with Michael Holden in Ep 1 of Protein Engineering in Practice (by @ranomics):
youtu.be/cVmGeFGsVA0
4
17
108
12,849