
Interpretable enzyme function prediction via sparse autoencoder features of ESMC across the microbial protein universe
1. The paper proposes a “features-first” route to enzyme function prediction: instead of training a new deep model, it uses ESMC-6B’s sparse autoencoder (SAE) features as an interpretable semantic signature to predict EC numbers.
2. Key enabler: the ESMC SAE expands layer-60 hidden states into a 16,384-feature codebook with Top-K=64 sparsity, and each feature has an independently interpretable biological concept label (generated/annotated via multi-agent GPT-5), enabling mechanistic explanations alongside predictions.
3. Benchmark setup: 4,868 reviewed microbial SwissProt enzymes (Bacteria Archaea), balanced across 7 EC1 superclasses and spanning 161 EC3 subclasses; proteins are 80–700 aa and have ≥3 EC levels. Protein representations are mean-pooled SAE activations, evaluated with simple linear probes.
4. Main EC3 result (161-way classification, 80/20 split): SAE binary features (just the top-64 active concepts per protein) reach 78.9% top-1 and 88.5% top-5 accuracy, outperforming a 3-mer logistic regression baseline (57.3% top-1) and approaching BLASTp (80.5% top-1).
5. Using richer SAE information improves further: the combined binary activation weights representation reaches 85.6% top-1 and 90.5% top-5, exceeding BLASTp top-1 by 6.4% in this benchmark.
6. Practical advantage over homology transfer: BLASTp returns no hit for 12.6% of test proteins, while SAE features yield predictions for 100% of queries; for the BLASTp no-hit subset, SAE binary still achieves 62.1% top-5 accuracy.
7. “Dark-matter regime” analysis: test proteins are binned by maximum 3-mer Jaccard similarity to training data; in the lowest-similarity bin (<0.20, containing 61% of the test set), SAE top-5 accuracy is 0.656 vs 0.438 for the 3-mer baseline, showing robustness when sequence similarity is minimal.
8. Generalization to unseen enzyme classes: leave-one-EC3-class-out evaluation (60 populous EC3 subclasses held out) trains only an EC1 classifier and tests whether it recovers the correct EC1 superclass for a completely unseen EC3 class; SAE binary achieves 47.7% EC1 recovery (3.3× random; vs 26.6% for sequence features).
9. Interpretability check: the most discriminative SAE features align with known enzymology—e.g., hydrolases linked to α/β-hydrolase catalytic triad/nucleophilic elbow geometry; oxidoreductases to Rossmann NAD(P)H-binding motifs; transferases to P-loop/Walker A phosphate-binding patterns; translocases to multi-helix transmembrane bundle concepts—supporting mechanistic plausibility of the learned “concept” features.
10. Atlas-scale survey: scanning 7.7M ESM Atlas cluster representatives, the authors identify 169,859 “dark enzyme-like” candidates (uncharacterized clusters with enzyme-suggestive Pfam keywords) spanning major microbial phyla, positioning SAE-based signatures as a scalable prioritization tool for experimental enzyme discovery.
💻Code: github.com/YueHuLab/esmc-sae…
📜Paper: arxiv.org/abs/2606.12209
#ProteinLanguageModels #EnzymeDiscovery #ECNumber #Interpretability #SparseAutoencoders #Microbiome #Metagenomics #ComputationalBiology #ESMC #ESMAtlas

5
34
2,229
Viral Proteins Reveal Geometry of Protein Language Models
1. The paper shows that protein language model (pLM) embedding spaces are dominated by a single “nativeness axis” (PC1) that strongly aligns with masked-reconstruction perplexity (a model-relative measure of how in-distribution a sequence is). This axis orders sequences from well-modeled cellular proteins to viral proteins to shuffled/random controls.
2. In ESMC-600M, PC1 explains 73.1% of embedding variance and correlates with perplexity at Spearman ρ=0.961, indicating that reconstruction difficulty is not just a score but a major geometric direction organizing embeddings across the tree of life.
3. Viral proteins are not treated as extreme outliers: they sit in an intermediate region—less “native” than cellular proteins but more structured than biologically meaningless sequences (position-shuffled or i.i.d. random), suggesting pLMs encode a continuum from in-distribution to out-of-distribution.
4. The same nativeness-axis geometry generalizes across ESM families (ESM2, ESMC, ESM3), including ESM3-OPEN (trained without viral sequences), and also appears in non-ESM architectures (ProGen2 autoregressive; EvoDiff discrete diffusion). This supports the idea that a dominant “model-fit” direction may be a broader property of sequence models trained on imbalanced biological data.
5. The work quantifies the underlying imbalance: UniRef50 pretraining coverage is heavily cellular-dominated (about 46.3M cellular clusters vs 390.3k viral; ~119× ratio), motivating the question of how underrepresented viral sequences are represented.
6. A key control argues nativeness is not simply “seen vs unseen”: cellular Swiss-Prot proteins released after an ESMC checkpoint (thus absent from its pretraining data) still look far more native-like (median PPL 5.3) than human viral proteins (median PPL 15.3), implying nativeness reflects compatibility with a cellular-dominated prior more than mere exposure.
7. Scaling changes viral nativeness, but unevenly across viral families: in ESMC (300M→6B), the fraction of human viral proteins below a native-like threshold (PPL<5) increases only modestly overall (~5%→~17%), while some families (e.g., Papillomaviridae, Retroviridae) shift strongly toward the native region and others (e.g., Orthomyxoviridae, Orthoherpesviridae, Sedoreoviridae) remain displaced.
8. Despite this dominant nativeness axis, embeddings retain viral-specific information beyond perplexity: linear probes on mean-pooled embeddings classify human viral vs cellular proteins with near-ceiling AUC (0.97–1.00 for larger models) under a homology-controlled split, and outperform both perplexity-only zero-shot classification and shallow sequence baselines (length, amino-acid composition, dipeptide composition).
9. The separation is especially relevant at low false-positive rates (screening-like settings): at 1% FPR, embedding probes achieve much higher TPR than perplexity-only classifiers (e.g., ESMC-6B 96.7% vs 39.2%), showing that “viral identity” is linearly accessible even when perplexity becomes a weaker separator at large scale.
10. Implications: nativeness (perplexity / PC1 position) can act as a diagnostic for where pLMs may be less reliable (notably for certain viral families), while embedding-based signals may complement homology methods for viral detection and biosecurity screening—though the authors emphasize evaluation and safety framing over deployment.
💻Code: github.com/MisteFr/viral-pro…
📜Paper: arxiv.org/abs/2606.12609
#ProteinLanguageModels #ESM #ViralProteins #RepresentationLearning #ComputationalBiology #Bioinformatics #MachineLearning #Biosecurity #Interpretability #ScalingLaws

2
41
3,170
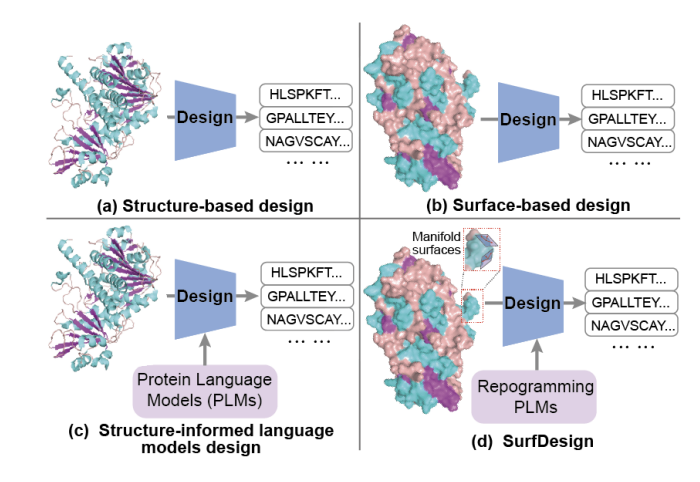
SurfDesign: Effective Protein Design on Molecular Surfaces
1. SurfDesign reframes protein design around molecular surfaces (shape physicochemical complementarity), aiming to better control functional regions like binding interfaces and enzyme pockets compared with backbone-only conditioning.
2. Core technical idea: treat molecular surfaces as continuous geometric manifolds rather than unordered point clouds/meshes, so the model can leverage local tangent structure, curvature, and directional consistency that are important for interaction-centric design.
3. The method builds an oriented surface point cloud Q where each point includes coordinates, a unit normal vector, and physicochemical attributes (e.g., hydrophobicity, charge, H-bond features). Surfaces are generated via PyMOL/MSMS and denoised with Gaussian smoothing; no residue identities, MSAs, or functional labels are used in surface generation.
4. SurfDesign introduces a Surface-conditioned Equivariant Message Passing (SEMP) encoder: SE(3)-equivariant updates use invariant radial distances, curvature descriptors (from local covariance eigenvalues), and directional angular features derived from surface normals (two intersection angles one dihedral angle).
5. Directionality is encoded with spherical Fourier–Bessel bases over distances and angles; messages are attention-reweighted and used to update both node features and coordinates, with optional per-layer recomputation of normals/curvatures to stay consistent as coordinates evolve.
6. To address limited surface-structure paired data and improve sequence priors, SurfDesign integrates pretrained protein language models (PLMs) via parameter-efficient fine-tuning (hybrid PEFT: structural adapter LoRA), trained with conditional masked language modeling rather than autoregressive decoding.
7. Binder design benchmark (6 targets) uses AF2 pAE_interaction as a functional proxy. SurfDesign achieves the best average pAE_interaction (15.85) and the highest overall success rate (30.14%), outperforming SurfPro (surface-conditioned baseline) and backbone-only baselines like ProteinMPNN, PiFold, and LM-DESIGN.
8. Enzyme design benchmark (5 enzyme–substrate systems; leakage-controlled by excluding overlaps with CATH pretraining) uses ESP score as a proxy for enzyme–substrate compatibility. SurfDesign attains the best average success rate (47.30%) and the best average ESP under greedy decoding (0.9058), with gains persisting in a zero-shot substrate setting.
9. Inverse folding is positioned as a diagnostic for structural compatibility (not “recovering the native sequence”). SurfDesign reports strong results on CATH splits, including perplexity 2.41 and AAR 74.13% on CATH 4.2, plus improved surface recovery metrics (IoU/CD/NC) versus PLM-based baselines; scaling larger PLMs further improves recovery.
💻Code: github.com/smiles724/SurfDes…
📜Paper: arxiv.org/abs/2606.07567
#ProteinDesign #ComputationalBiology #GeometricDeepLearning #ProteinEngineering #EnzymeDesign #ProteinBinding #EquivariantNetworks #ProteinLanguageModels #KDD2026
15
74
3,913
Heuristic multi-site optimization for protein sequence design using Masked Protein Language Models @PLOSCompBiol
1 ProtHMSO is a heuristic protein sequence design framework that uses masked protein language models (mainly ESM-2) to propose context-aware, multi-site substitutions, aiming to escape local optima and reduce the “invalid/destabilizing” variants common in blind random mutagenesis.
2 Key idea: mask one or multiple target positions, let the ProtLM output substitution probabilities conditioned on the entire sequence context, and use top-k (k=3 worked best) candidate substitutions to generate a small, high-potential mutant set for fitness scoring—shrinking combinatorial search while keeping evolutionary/biophysical plausibility.
3 The multi-site masking is central: substitutions for all masked sites are predicted synchronously from global context, so probabilities update as the sequence changes. This provides a zero-shot way to capture epistasis (synergistic residue interactions) without explicit structural supervision or task-specific training.
4 ProtHMSO is positioned as both (a) a standalone iterative optimizer and (b) a plug-in mutation operator that can replace random exploration steps inside classic search methods, improving convergence and sample-efficiency.
5 GA-HMSO: integrates ProtHMSO into a genetic algorithm by replacing random mutation with ESM-2-guided mutation, and uses a multi-objective fitness (sum of predictor scores). A dynamic schedule (higher mutation early, lower later) improved exploration–exploitation balance and avoided premature convergence.
6 MCTS-HMSO: integrates ProtHMSO into Monte Carlo Tree Search by using ESM-2 probabilities to guide expansion. It also introduces grouping of child nodes by mutation position (choose site first, then substitution), mitigating the wide-and-shallow tree problem in high-dimensional sequence action spaces.
7 AMP benchmark (DBAASP-derived; three challenging cases): across 1–5 site mutations, ProtHMSO consistently improved antimicrobial metrics (PAMP, PMIC) over random mutagenesis, while also improving structural plausibility proxies (higher ESMFold pLDDT, lower ProGen2 perplexity). Notably, random multi-site mutation degraded plausibility as sites increased, while ProtHMSO improved it.
8 ProteinGym benchmark (long proteins; single-site focus for scalability): on the Clinical substitution benchmark, ProtHMSO produced about 2x more non-pathogenic variants than random mutation at matched library sizes (10/50/100 variants per sequence). On DMS benchmarks (including targeted functional-site mutagenesis), ProtHMSO showed higher enrichment of experimentally top-ranked high-fitness mutants (top-10/20/50 overlaps) for both single- and two-site settings.
9 Practical framing: ProtHMSO acts as a high-throughput “candidate narrowing” layer—reducing millions of possibilities to tens/hundreds—while remaining compatible with adding stricter downstream filters (e.g., Rosetta/MD) in a coarse-to-fine pipeline.
💻Code: github.com/chen-bioinfo/Prot…
📜Paper: doi.org/10.1371/journal.pcbi…
#ProteinDesign #ProteinEngineering #ProteinLanguageModels #ESM2 #DirectedEvolution #GeneticAlgorithms #MCTS #AntimicrobialPeptides #ProteinGym #ComputationalBiology

1
25
2,046
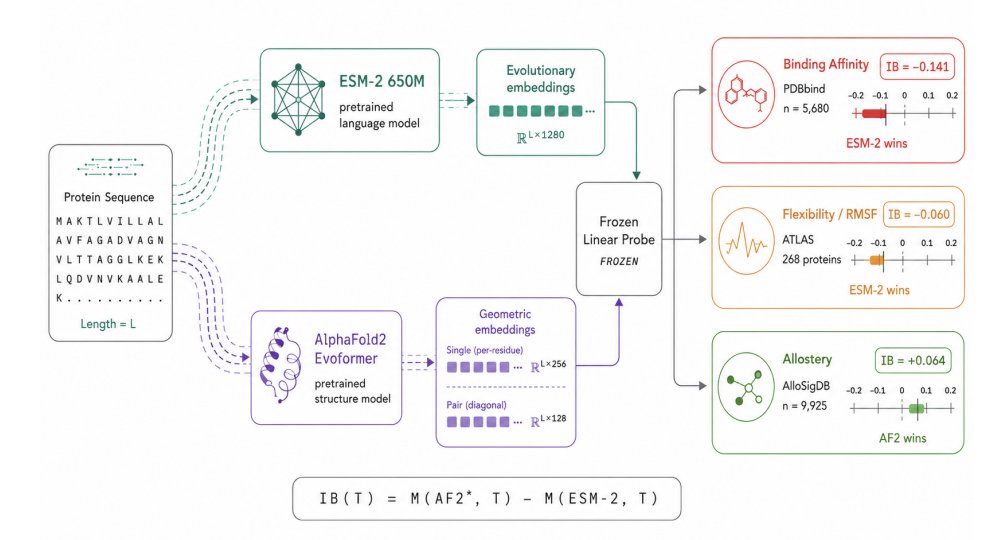
When Does Structure Help? The Information Bonus of AlphaFold2 Representations over Protein Language Models
1. The paper introduces Information Bonus (IB): a task-level metric that quantifies how much linearly accessible signal is gained by using frozen AlphaFold2 (AF2) Evoformer representations instead of a cheaper frozen sequence-only model (ESM-2), evaluated under protein-level cross-validation.
2. IB is defined as the held-out performance difference between the best AF2 representation (chosen post-hoc between Evoformer single vs pair-diagonal) and ESM-2, using the same frozen linear probe. IB > 0 means structure adds usable signal; IB < 0 means sequence embeddings are sufficient or better.
3. The most decisive positive-IB regime is allostery (AlloSigDB; 47 proteins, 9,925 residues, 4.8% positives): AF2 single achieves AUROC 0.548, while ESM-2 is below chance at 0.485 and AF2 pair-diagonal is near chance at 0.497. This suggests AF2 single encodes long-range geometric/communication-network information that is not linearly recovered from sequence alone.
4. Binding affinity (PDBbind; n=5,680 complexes) shows a strong negative IB: ESM-2 reaches Pearson r=0.449 vs AF2 single r=0.307 and AF2 pair-diagonal r=0.278 (IB = -0.141). The paper argues this likely reflects evolutionary/family-level binding constraints captured by sequence models.
5. A key experimental design choice: the affinity probe receives only protein features (no ligand representation). So the benchmark tests whether representations capture protein-level correlates of affinity (e.g., pocket druggability, family propensity), not ligand-specific complementarity; AF2 features also reflect an apo-like inference rather than the bound complex.
6. Flexibility (ATLAS MD; 268 proteins, 50,426 residues) is mixed and label-dependent. For RMSF regression, AF2 pair-diagonal is directionally best (r=0.436) vs ESM-2 (r=0.407), giving a small positive IB ( 0.030) with limited statistical power across 5 folds.
7. For within-protein median flexibility classification, ESM-2 wins clearly: AUROC 0.824 vs AF2 pair-diagonal 0.764 and AF2 single 0.762 (IB = -0.060; p=0.0017 vs AF2 pair). Interpretation: sequence context captures disorder/mobility signatures better than static geometry for this relative-flexibility label.
8. The paper highlights a residue-level leakage artifact: naive residue-wise KFold (allowing residues from the same protein in both train/test) inflates RMSF performance by 27–39% depending on representation (e.g., ESM-2 r=0.672 under leaky split vs 0.407 under protein-level GroupKFold). This inflation can reverse representation rankings and change conclusions.
9. Practical takeaway framed for AI-scientist workflows: representation choice should be a measurable decision. Start with ESM-2 when labels are plausibly driven by evolutionary constraints or disorder-like sequence signatures; pay the AF2 inference cost when the mechanism depends on long-range 3D communication (as in allostery). When uncertain, estimate IB on a small labeled set before scaling structural inference.
📜Paper: arxiv.org/abs/2606.04228
#ComputationalBiology #ProteinML #AlphaFold2 #ProteinLanguageModels #RepresentationLearning #Allostery #Benchmarking #DataLeakage #AIFORScience
5
32
2,271
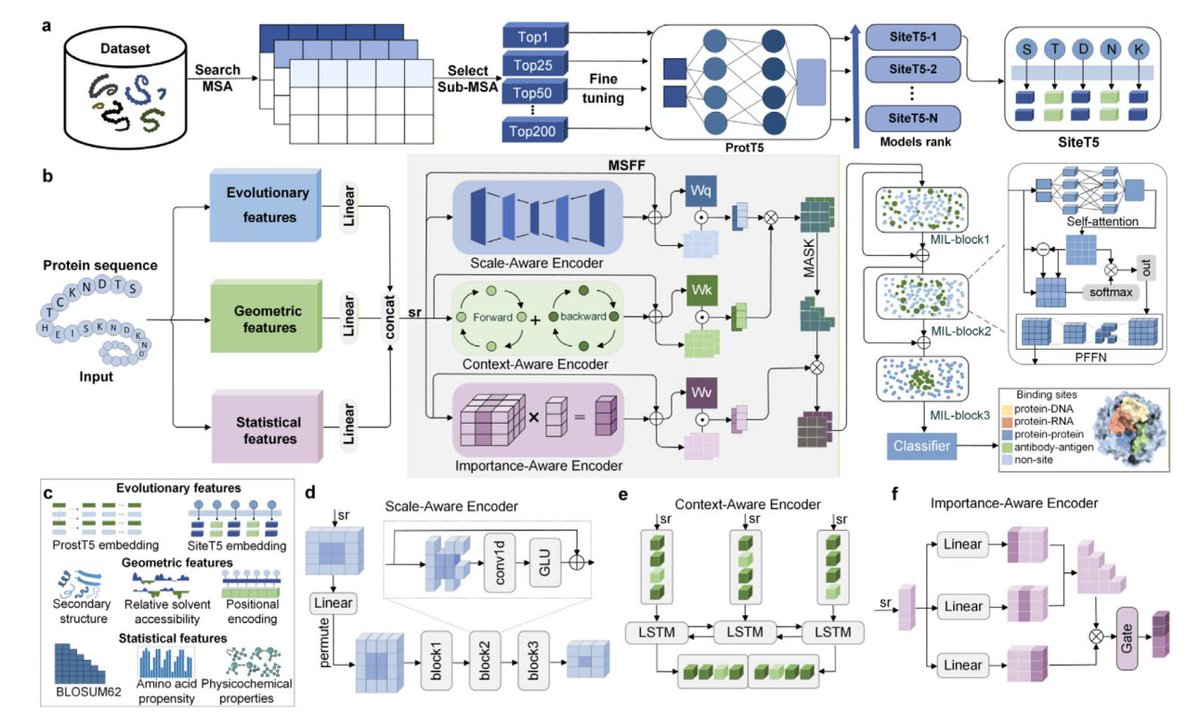
ProSiteHunter: A Unified Framework for Sequence-Based Prediction of Protein-Nucleic Acid and Protein-Protein Binding Sites
1. ProSiteHunter is presented as a unified, sequence-only framework that predicts residue-level binding sites across four interaction types: protein-DNA, protein-RNA, protein-protein, and antibody-antigen, aiming to replace the usual “one model per interface type” fragmentation in sequence-based predictors.
2. The core idea is multi-source sequence representation: it combines (i) a task-specific fine-tuned protein language model SiteT5 for evolutionary/functional-site signals, (ii) ProstT5 embeddings for structure-related priors, plus (iii) geometric descriptors derived from sequence-predicted properties (secondary structure, relative solvent accessibility, symmetric position encoding), and (iv) statistical descriptors (BLOSUM62, physicochemical properties, amino-acid propensity).
3. A key architectural contribution is the Multi-Source Feature Fusion (MSFF) module with “three-track semantic parsing”: Scale-Aware Encoder (multi-kernel 1D CNNs for local patterns), Context-Aware Encoder (BiLSTM for bidirectional semantics), and Importance-Aware Encoder (gated self-attention for long-range dependencies). These tracks are mapped into Q/K/V and fused via cross-attention for dynamic alignment across feature spaces.
4. A second stage, Multi-Level Interaction Learning (MIL), stacks gated multi-head self-attention blocks plus position-wise feed-forward networks to iteratively refine interface signals, producing per-residue binding probabilities (thresholded at 0.5 for site calls).
5. SiteT5 is introduced as a task-adapted PLM derived from ProtT5-XL-UniRef50, fine-tuned with evolutionary information from sub-MSAs (generated with HHblits on UniRef30). Fine-tuning uses LoRA and updates only the last four decoder layers, yielding a relatively small number of trainable parameters while specializing to binding-site patterns.
6. On GraphBind-style temporal splits for nucleic-acid interfaces, ProSiteHunter reports strong gains over prior sequence methods (e.g., CLAPE variants, iDRNA-ITF, DRNApred), emphasizing PRAUC improvements under heavy class imbalance (site:non-site ≈ 1:10), alongside higher ROCAUC/F1/MCC.
7. On protein-protein binding sites (Seq-InSite dataset) and antibody-antigen epitopes (SEMA conformational epitope dataset), the same unified design remains competitive, reporting improvements over methods such as Seq-InSite/ISPRED-SEQ for PPI and CALIBER for antibody-antigen, with particularly notable PRAUC gains on the epitope task.
8. The paper positions ProSiteHunter as complementary to structure predictors: it highlights cases where structure-based approaches (including AlphaFold3) can mis-localize interfaces when structures are imperfect or when binding involves flexible regions, while sequence-driven predictions remain stable and can flag “local flexible sites.”
9. Ablations support the design rationale: removing SiteT5 or ProstT5 embeddings causes the largest drops (SiteT5 removal being most damaging), while removing geometric/statistical features yields smaller but consistent degradations; removing MSFF or MIL leads to substantial performance loss, with MSFF identified as the larger contributor.
10. A downstream demonstration integrates ProSiteHunter-predicted epitopes into an in-house antibody-antigen interaction predictor (Multi-sAAI), reporting improved interaction classification metrics (ROCAUC/F1/precision/recall) and case studies where predicted epitope features sharply increase predicted interaction probabilities for known therapeutic or broadly neutralizing antibody scenarios.
📜Paper: doi.org/10.1002/advs.75931
#ComputationalBiology #Bioinformatics #ProteinScience #ProteinLanguageModels #DeepLearning #ProteinInteractions #EpitopePrediction #PPI #ProteinDNA #ProteinRNA #AntibodyEngineering #DrugDiscovery
4
21
1,887
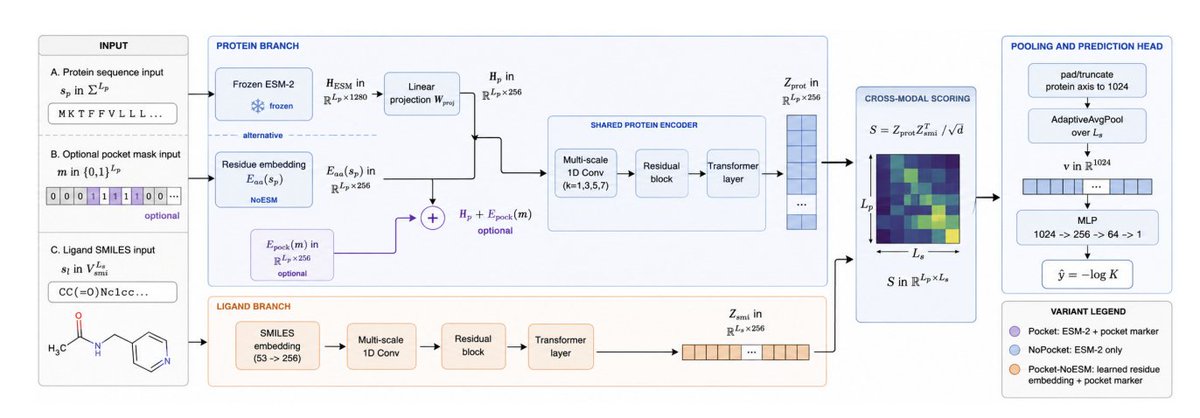
HonestAffinity: Leak-Aware Evaluation of Protein and Pocket Priors for Binding Affinity Prediction
1. HonestAffinity frames a key caution for protein–ligand affinity models: architectural “priors” can flip from helpful to harmful depending on whether evaluation splits leak protein/ligand similarity (canonical CASF/PDBbind-style) or are leak-proof (LP-PDBBind 3-tier no-leak).
2. The paper isolates two common priors in a controlled 1D-input setting: frozen ESM-2 (650M) per-residue embeddings (1280-d projected to 256) and a learned binary pocket-position marker added to residue features when pocket annotations are available.
3. Core result is a split-conditioned reversal across both priors. On familiar/canonical splits (val, CASF-2016, CASF-2016 non-train), adding ESM-2 and the pocket marker improves performance; on strict LP no-leak tiers (test_cl1–cl3), the same additions reduce Pearson R.
4. Three deployment-matched variants are proposed rather than one “best” model: HONESTAFFINITY-POCKET (ESM-2 pocket marker) for familiar/annotated targets; HONESTAFFINITY-NOPOCKET (ESM-2 only) when no pocket list exists; HONESTAFFINITY-POCKET-NOESM (21-token residue embedding pocket marker) for strict LP-style generalization with pocket annotations.
5. Quantitatively (Pearson R, mean±std over 3 seeds): HONESTAFFINITY-POCKET leads on val (0.548), CASF-2016 (0.747), and CASF non-train (0.646). But on LP strict tiers, HONESTAFFINITY-POCKET-NOESM leads: cl1 0.531, cl2 0.538, cl3 0.497, also giving best RMSE on cl2/cl3.
6. The ESM-2 ablation is especially instructive: swapping ESM-2 for a learned 21-vocab residue embedding decreases R on val/CASF (e.g., CASF-2016 drops from 0.747 to 0.713) but increases R on every strict LP tier (e.g., cl3 rises from 0.433 to 0.497; ∆R up to 0.064).
7. The pocket marker shows the same sign flip: it helps on val/CASF but hurts on LP tiers. Interpretation: both priors inject signals correlated with training-distribution structure (protein-family signatures or pocket geometry), which can become misleading when similarity filtering removes overlap by design.
8. Methodologically, the authors argue for paired reporting: canonical metrics alone would over-credit ESM/pocket priors; leak-proof metrics alone would understate their utility for in-distribution scoring. They recommend routine paired canonical leak-proof ablations for new affinity predictors.
9. Implementation is intentionally compact and scalable: multi-scale 1D CNNs a residual block a single Transformer layer per branch (protein and SMILES), coupled by a matrix-product compatibility map. Training uses 11,513 LP-PDBBind train complexes and runs in ~3 GPU-hours on a single V100; inference is ~10 ms/complex with cached embeddings.
📜Paper: arxiv.org/abs/2606.03422
#CompBio #Bioinformatics #DrugDiscovery #ProteinLanguageModels #ESM2 #BindingAffinity #Benchmarking #PDBbind #CASF2016 #MachineLearning
5
19
1,588

May 29
Excited to share our #RECOMB2026 work, DETANGO: a deep learning framework for disentangling mutation effects on protein stability and function from evolutionary signals captured by protein language models (pLMs).
DETANGO estimates a functional plausibility score that quantifies mutation effects on function beyond what can be explained by stability alone. Across extensive benchmarks, DETANGO accurately identifies stable-but-inactive (SBI) variants and functionally important residues involved in ligand binding, catalysis, and allostery.
Grateful to my co-authors @ZiangLi2001, @Qwe1029384756Tu, and Jiaqi Luo, and to my advisor @luoyunan for their invaluable contributions and support throughout this work! Special thanks to Tony for representing our team and presenting DETANGO today at RECOMB 2026 in Thessaloniki, Greece!
Preprint: biorxiv.org/content/10.64898…
Code: github.com/luo-group/DETANGO
#MutationEffectPrediction #ProteinLanguageModels #ProteinFunction #ComputationalBiology #DeepLearning
1
2
269
ProteomeLM: A proteome-scale language model enables accurate and rapid prediction of protein–protein interactions and gene essentiality across taxa
1. ProteomeLM is a transformer that operates on entire proteomes (not single proteins or local genomic windows), learning organism-level constraints that help predict protein–protein interactions (PPI) and gene essentiality across the tree of life.
2. Key idea: represent each protein by a pretrained sequence embedding (ESM-Cambrian, 1152D averaged over residues), then train ProteomeLM with a proteome-level masked objective: mask many proteins and reconstruct their embeddings using the remaining proteins in the same proteome.
3. A major design choice is dropping genomic positional encoding (problematic across taxa, especially eukaryotes) and instead adding a functional encoding derived from OrthoDB orthologous groups, sampled at multiple taxonomic levels to expose the model to coarse-to-fine functional identity during training.
4. Technical novelty in training: a custom “polar loss” for reconstructing continuous embeddings, designed to avoid a degenerate solution where the model collapses to reproducing the functional encoding; it separately penalizes residual direction (cosine term) and magnitude (squared norm difference).
5. Unsupervised PPI signal emerges in attention: without any interaction labels, specific attention heads strongly separate interacting vs noninteracting pairs across species (e.g., AUC up to ~0.92 in E. coli for a single head; strong cross-species heads also observed), with PPI signal enriched in intermediate layers.
6. The attention signal captures multiple relationship types: direct physical binding, same-complex membership, and broader genetic/functional associations (e.g., coexpression). It tends to recover broad functional associations and complex membership particularly well, and can partially disentangle physical vs functional relationships.
7. Interactome-wide screening: a lightweight logistic regression on ProteomeLM attention heads (trained with only ~100 positives 1,000 random negatives) enables proteome-scale PPI ranking that is far faster than DCA pipelines and more accurate in large screens (e.g., human AUC ~0.83 vs ~0.73 reported for DCA; substantially higher recall among top-ranked pairs).
8. Compute advantage: once ProteomeLM is trained, inference (including ESM-C embeddings attention extraction) is reported to take under ~10 minutes per proteome (e.g., human) on a single GPU, avoiding per-pair MSA building and per-pair model fitting required by DCA; the paper estimates up to ~6 orders of magnitude speedup for inference in human-scale screens.
9. Supervised PPI prediction (ProteomeLM-PPI): a downstream network that combines node features (ESM-C ProteomeLM embeddings) and edge features (attention coefficients) reaches state-of-the-art or comparable performance across benchmarks, with especially strong cross-species generalization (notably large AUPR gains on E. coli when trained on human PPI).
10. Gene essentiality (ProteomeLM-Ess): a simple supervised classifier on ProteomeLM embeddings improves over using protein-only ESM-C embeddings, reaching strong performance across 91 species (OGEE) and generalizing to held-out proteomes and even minimal/synthetic cells (JCVI-Syn1.0, JCVI-Syn3A), with best reported AUC up to ~0.93 depending on model size/layer.
💻Code: github.com/Bitbol-Lab/Proteo…
📜Paper: doi.org/10.1073/pnas.2524201…
#ComputationalBiology #Bioinformatics #ProteinInteractions #PPI #ProteinLanguageModels #Transformers #SystemsBiology #GeneEssentiality #FoundationModels #Proteomics
7
43
2,364
Scaling antibody language models improves structure aware representation for antibody engineering
1 AbLingua introduces a structure-aware way to pre-train antibody language models by changing the basic “unit” of meaning from single amino acids to overlapping tripeptides, aiming to better capture local cooperative interactions that drive folding and CDR conformations.
2 The work scales an antibody-specific encoder (BERT-style) up to 1.7B parameters, trained on 1.4B antibody sequences from OAS (unpaired; ~1.2B heavy and ~200M light), positioning it as a large encoder-based foundation model specialized for antibodies.
3 Core technical contribution: TripleAA tokenization. It converts sequences into overlapping 3-residue tokens with boundary markers, expanding the vocabulary from 20 amino acids to 21,952 fixed-length tokens, avoiding variable-length “label island” issues and making residue-level supervision easier to align.
4 Pre-training is modified with Context Residue Transformation (CRT): (i) joint mask masks all tokens containing a chosen central residue to prevent leakage from overlapping tripeptides, and (ii) joint replacement replaces the central residue consistently across overlapping tokens while sampling replacements from natural amino-acid frequencies.
5 The paper reports predictable scaling behavior for antibody LMs when using this tokenization: increasing parameters and data volume reduces perplexity following power-law-like trends, supporting the idea that antibody LMs can benefit from scaling when the encoding granularity matches biology.
6 Paratope prediction: AbLingua-1.7B is evaluated for unified prediction across both CDR and framework (FR) residues, addressing strong class imbalance (FR paratopes are rare). Fine-tuned performance is reported around 72% recall and 75% F1, outperforming multiple antibody/protein LMs and surpassing an MSA-based specialized baseline after full fine-tuning.
7 A structural case study (RSV complex PDB 6apb) suggests attention concentrates on known binding residues within CDRH3 (not just broadly on the loop), indicating the pre-trained representations can localize functionally relevant sites without explicit paratope supervision during pre-training.
8 Neutralization prediction across SARS-CoV variants: using CoV-AbDab-derived datasets (11,008 sequences; separated by epitope region NTD/RBD and variant), AbLingua-1.7B yields more stable AUROC across variants than general protein LMs, with performance dropping most on highly mutated Omicron BA.1/BA.2 yet remaining usable (reported AUROC ~65% for the hardest cases).
9 Therapeutic discovery setting (HER2 CDRH3 binder prediction): combining AbLingua embeddings with a downstream ResNet improves recall on noisy high-throughput labels and generalizes to an independent gold-standard BLI-tested set; validation recall is reported at 58%, and test recall up to 85% with more training data, with robustness across differing numbers of CDRH3 mutation positions.
10 Unsupervised analyses: AbLingua embeddings better separate (via UMAP) B-cell developmental stages and virus-specific antibody groups (HIV/Ebola/SARS) without labels, and token-level projections show more context-sensitive representations for frequent residues compared with baselines, consistent with the intended structural-awareness of the tripeptide vocabulary.
📜Paper: doi.org/10.1038/s42003-026-1…
#AntibodyEngineering #ProteinLanguageModels #ComputationalBiology #Immunoinformatics #MachineLearning #DeepLearning #Therapeutics #BERT #ScalingLaws #Bioinformatics
4
11
1,398
EvoStruct: Bridging Evolutionary and Structural Priors for Antibody CDR Design via Protein Language Model Adaptation
1. EvoStruct addresses a practical failure in structure-conditioned antibody CDR design: equivariant GNNs can score high on sequence recovery but collapse their outputs to a tiny amino-acid vocabulary (often overpredicting Tyr/Gly and missing rare but important residues like Trp/Cys/Met).
2. The paper traces vocabulary collapse to a root cause: GNN encoders must learn amino-acid distributions de novo from limited structural complexes (~3k), effectively discarding evolutionary substitution patterns that protein language models (PLMs) already encode from hundreds of millions of sequences.
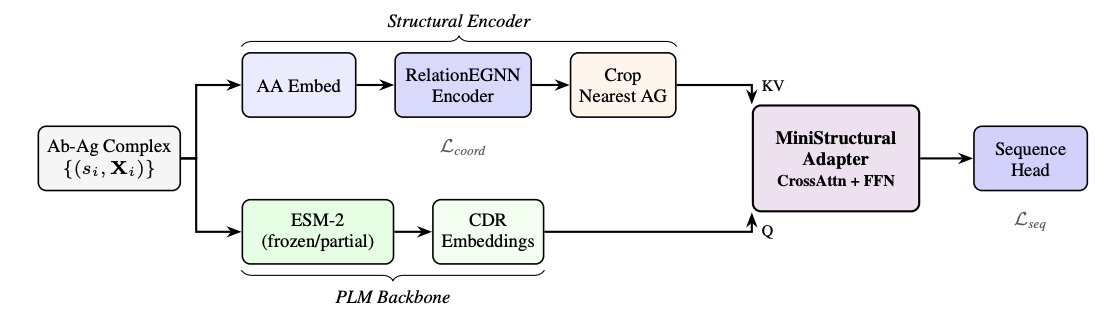
3. Core idea: keep sequence prediction in a PLM’s calibrated embedding space while injecting 3D context. EvoStruct bridges frozen/partially-frozen ESM-2 with an E(3)-equivariant relation-aware GNN using a cross-attention “mini structural adapter” operating in ESM-2 representation space.
4. Architectural separation of concerns: the GNN focuses on geometry of the antibody–antigen complex (typed edges, equivariant coordinate updates), while ESM-2 provides evolutionary priors and well-calibrated amino-acid probabilities; cross-attention lets structural context modulate PLM representations without overwriting them.
5. Training specifically targets CDR design pathologies (not just generic inverse folding): progressive PLM unfreezing to avoid catastrophic forgetting of evolutionary priors, plus R-Drop consistency regularization to stabilize predictions under dropout and encourage robust context injection.
6. The study introduces/uses diagnostics to quantify collapse and conditioning: effective vocabulary (Veff, exponentiated entropy) and paratope–epitope amino-acid pair correlation, alongside standard metrics like AAR/CAAR, perplexity, RMSD, fnat, DockQ, epitope F1, and sequence liabilities.
7. On CHIMERA-BENCH (epitope-group split; 2,922 SAbDab complexes), EvoStruct ranks best on sequence metrics: AAR 0.43 (vs 0.37 best GNN baselines; 16% relative) and perplexity 1.88 (vs 3.27 RAAD; -43%), while maintaining comparable backbone RMSD (~1.84 Å).
8. EvoStruct substantially reduces vocabulary collapse: Veff = 12.4 (~80% of native diversity) vs GNN greedy-decoding baselines Veff ~4.7–5.3 (~30–34% of native). It also recovers far more motifs (e.g., 282 unique bigrams and 1,214 trigrams vs RAAD 52/110), indicating improved diversity without sacrificing accuracy.
9. Conditioning improves where baselines struggle: EvoStruct shows the strongest binding-pair learning (paratope–epitope pair correlation r = 0.73; interface enrichment correlation r = 0.81), suggesting cross-attention successfully routes antigen information to the sequence head, though contact-position recovery remains difficult for all methods.
10. Limitations noted: EvoStruct does not lead binding-quality metrics (e.g., RefineGNN—without antigen conditioning—still tops fnat/DockQ), and contact AAR remains low (~22%), implying future work may need richer antigen-conditioning mechanisms or objectives tailored to interface positions.
📜Paper: arxiv.org/abs/2605.21485
#ComputationalBiology #AntibodyDesign #ProteinDesign #ProteinLanguageModels #GeometricDeepLearning #GNN #ESM2 #InverseFolding #CDR #MachineLearning
4
15
1,876
Advancing Knotted Protein Design with ESM3: Guided Generation and Topological Insights
1. The study probes whether multimodal protein language models capture topology by using knotted proteins (rare <1% of structures) as a stringent test case, focusing on the smallest open ESM3 variant (ESM3-SM, 1.4B).
2. Using derivative-free guided decoding with a topology-aware objective (Topoly Alexander polynomial via stochastic closure), ESM3 generates knotted proteins de novo from a fully masked 256-aa input with 89% success (89/100; 95% CI 81–94), far above prior unguided diffusion-based reports (~0.5%, noting the comparison is guided vs unguided).
3. Generated knots are diverse: among resolved cases, trefoils (3_1) dominate (33), with figure-eight (4_1) and torus (5_2) also frequent; the model also produces more complex knots up to 9 crossings, while ~21% exceed the Alexander-polynomial default crossing limit and remain unclassified.
4. Knot-type targeting is possible by modifying the scoring function: targeting 3_1 works well (9/10), while more complex targets (4_1, 5_1, 5_2) are achieved ~20–30% and otherwise tend to fall back to the most probable topology (often 3_1).
5. Length strongly affects success: generation is unreliable at ~100 aa (20% knotted), improves by 200 aa (~90%), and reaches ~100% at 350 aa , matching the typical 200–400 aa range of natural knotted proteins.
6. Topology is strikingly robust to sequence perturbation: with randomized masking ESM3 regeneration, 94% of knotted proteins remain knotted at 50% masking, and 85% remain knotted at 70% masking; the mean “breaking point” is ~84% sequence alteration (±1.2% SE).
7. Knot loss shows threshold-like behavior rather than gradual decay: many proteins exhibit abrupt drops in knot probability between consecutive masking levels, consistent with a critical amount of distributed/redundant sequence information being sufficient to specify topology.
8. Structural drift accumulates well before topological disruption: at 50% masking, median RMSD is already ~3.24 Å (sequence identity ~73%) while mean knot probability stays ~0.85; at 70% masking RMSD ~4.58 Å while knot probability ~0.76, suggesting topology is more robust than precise 3D coordinates.
9. Multiple analyses argue against simple memorization: de novo generation starts from all-mask inputs; generated sequences have very low similarity to known knotted proteins (max 14.5%, mean 11.6%, comparable to random baseline); and topology can persist despite large sequence changes.
10. The work also demonstrates partial topological “editing”: iterative low-rate masking with guidance converts 17/99 short unknotted proteins (≤250 aa) into knotted variants (17% success; 95% CI 10–26%), with a bimodal pattern (some convert quickly, many resist conversion).
💻Code: github.com/ML-Bioinfo-CEITEC…
📜Paper: biorxiv.org/content/10.64898…
#ProteinDesign #GenerativeAI #ProteinLanguageModels #ESM3 #Topology #KnottedProteins #ComputationalBiology #Bioinformatics #Biosecurity

7
51
3,098
Learning the PTM Code through a Coarse-to-Fine Mechanism-Aware Framework
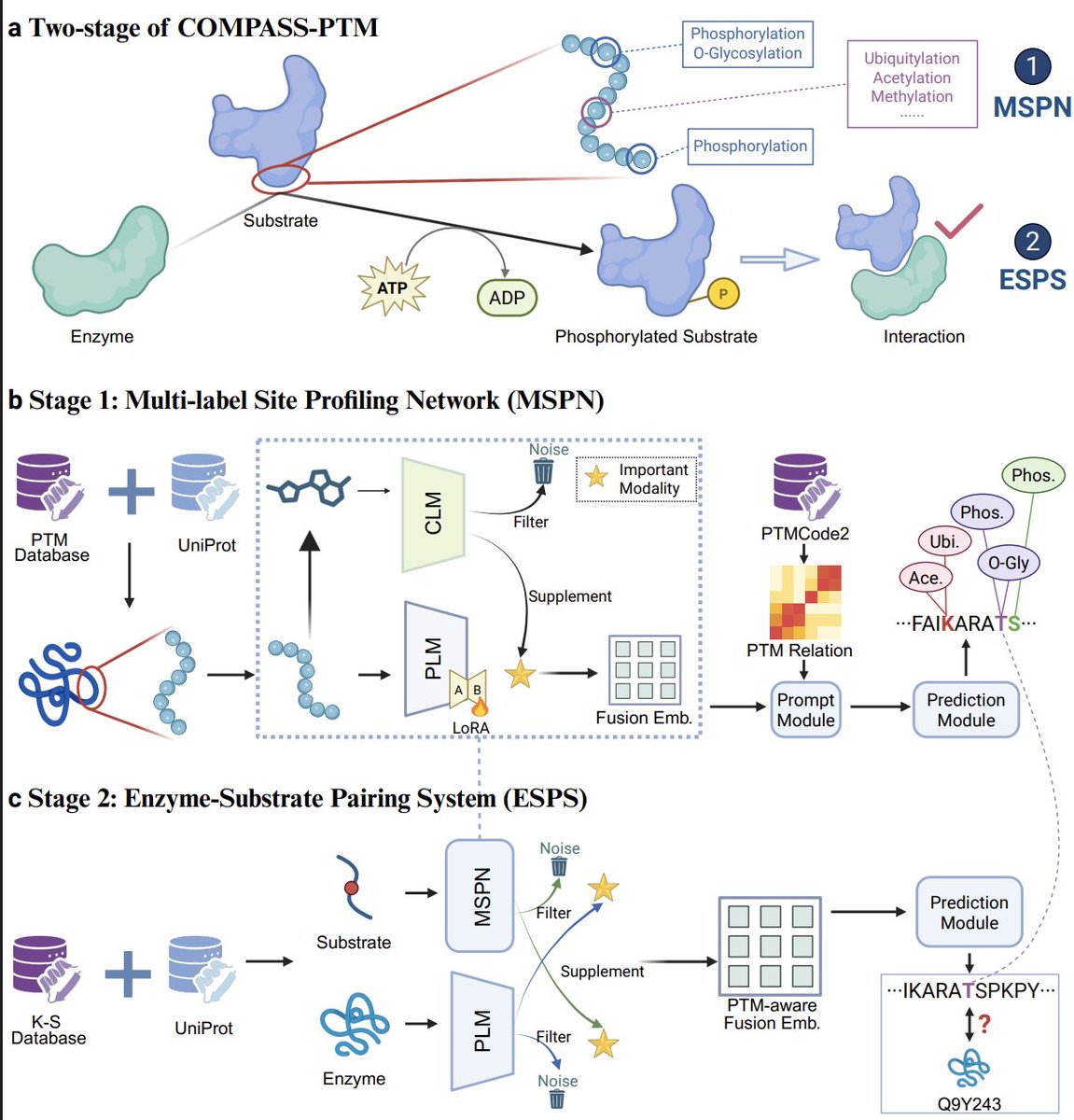
1. The paper introduces COMPASS-PTM, a two-stage framework that jointly learns residue-level multi-label PTM site profiles and enzyme–substrate assignment, framing them as two resolutions (coarse-to-fine) of the same underlying PTM “regulatory program.”
2. Stage 1 (MSPN) targets realistic PTM complexity: multiple PTM types can co-occur at the same residue, and predictions should remain biologically coherent rather than treating each PTM/site independently.
3. A key idea is crosstalk-aware prompting: a learnable PTM-type × PTM-type prior matrix (initialized from empirical PTM co-occurrence) is injected as an attention bias, encouraging predictions that reflect cooperative/antagonistic PTM relationships and reducing implausible multi-label outputs.
4. MSPN uses a dual-modal encoder: a protein language model backbone (ESM2-150M) for evolutionary/sequence context plus a chemical modality (SELFIES-based chemical language model embeddings physicochemical descriptors) to capture residue reactivity; fusion is explicitly designed to keep PLM features primary while selectively augmenting with chemical signals.
5. The training objective explicitly addresses PTM’s “dual long-tail” imbalance: rare PTM types across classes and extreme sparsity of modified residues across positions. A hybrid loss (macro regularized Dice micro focal, with additional regularization against over-confidence) is used to improve rare-class learning while controlling false positives.
6. On multi-label proteome-scale benchmarks curated from dbPTM and qPTM, MSPN reports large gains over evaluated baselines, including a 122% relative improvement in macro-F1 for multi-label site prediction; the paper emphasizes that improvements are driven by better precision (practical for experimental follow-up), not just AUROC.
7. Beyond per-residue metrics, COMPASS-PTM reduces “spurious multi-label burden” (over-calling extra PTM types once a site is detected), producing cleaner PTM label sets and higher protein-level “program” coherence.
8. Stage 2 (ESPS) reuses the PTM-aware substrate representation from Stage 1 and refines it into enzyme-resolved hypotheses by learning enzyme embeddings and predicting enzyme–substrate relationships; evaluation includes warm-start and stringent cold-start splits for unseen substrates or unseen enzymes.
9. ESPS improves enzyme assignment on OmniPath and SAGEPhos benchmarks, and achieves strong generalization in a true zero-shot setting on DARKIN (disjoint kinases between train/test), reporting a 54% mAP gain over the previously best reported score.
10. The framework is positioned as interpretable and variant-aware: embeddings organize into biochemically meaningful clusters; recovered kinase-family motifs match canonical consensus patterns; and case studies link missense variants to both local PTM gain/loss and predicted rewiring of enzyme–substrate interactions (e.g., LRRK2 variants affecting PKA-linked phosphorylation; hypotheses for SCNN1B and FUS).
💻Code: github.com/ZhangJJ26/COMPASS…
📜Paper: doi.org/10.1038/s41467-026-7…
#ComputationalBiology #Bioinformatics #Proteomics #PostTranslationalModifications #ProteinLanguageModels #DeepLearning #SystemsBiology #Kinase #Ubiquitination #VariantInterpretation
5
25
2,413
Deep Learning for Protein Complex Prediction and Design
1. Ziwei Xie’s 2026 PhD thesis connects two directions that are often treated separately: predicting protein complex structures (forward problem) and designing protein binders on fixed backbones (inverse problem), emphasizing domain-specific neural architectures plus search/pairing strategies.
2. A central practical bottleneck addressed is heterodimer prediction quality being limited by how well one can identify true interacting homolog pairs (interologs) across species; the thesis argues that better pairing can directly translate into better AlphaFold-Multimer inputs and better complex models.
3. GLINTER: a supervised interface contact predictor that fuses (i) monomer structural graphs and (ii) co-evolutionary signals extracted as attention maps from an MSA Transformer (ESM-MSA). It is designed to work with either experimental monomer structures or predicted monomers (e.g., AlphaFold).
4. GLINTER’s architecture is explicitly geometry-aware: a Siamese graph-convolution module processes multiple monomer-derived graphs (residue graph, atom graph, surface graph) with local reference frames for rotation-invariant message passing, then a ResNet consumes the pairwise outer-concatenated monomer embeddings plus inter-chain attention features to output interfacial contact probabilities.
5. Beyond contact-map metrics, GLINTER is positioned as a docking aid: predicted top interfacial contacts are used to rank/select docking decoys (e.g., from HDOCK), improving the average quality of selected decoys, highlighting contact prediction as a practical intermediate for complex modeling pipelines.
6. ESMPair: a protein-language-model-enhanced MSA pairing pipeline for heterodimers. It uses column-wise attention scores from pretrained ESM-MSA-1b to rank homologs per chain within each species, then pairs same-rank homologs to create interolog MSAs for AlphaFold-Multimer.
7. Reported outcomes for ESMPair focus on heterodimers (especially eukaryotic targets) where phylogeny/genome-based pairing is error-prone; ESMPair improves DockQ and success rate, and ensemble strategies (e.g., ESMPair genome pairing AF-Multimer defaults) further increase robustness.
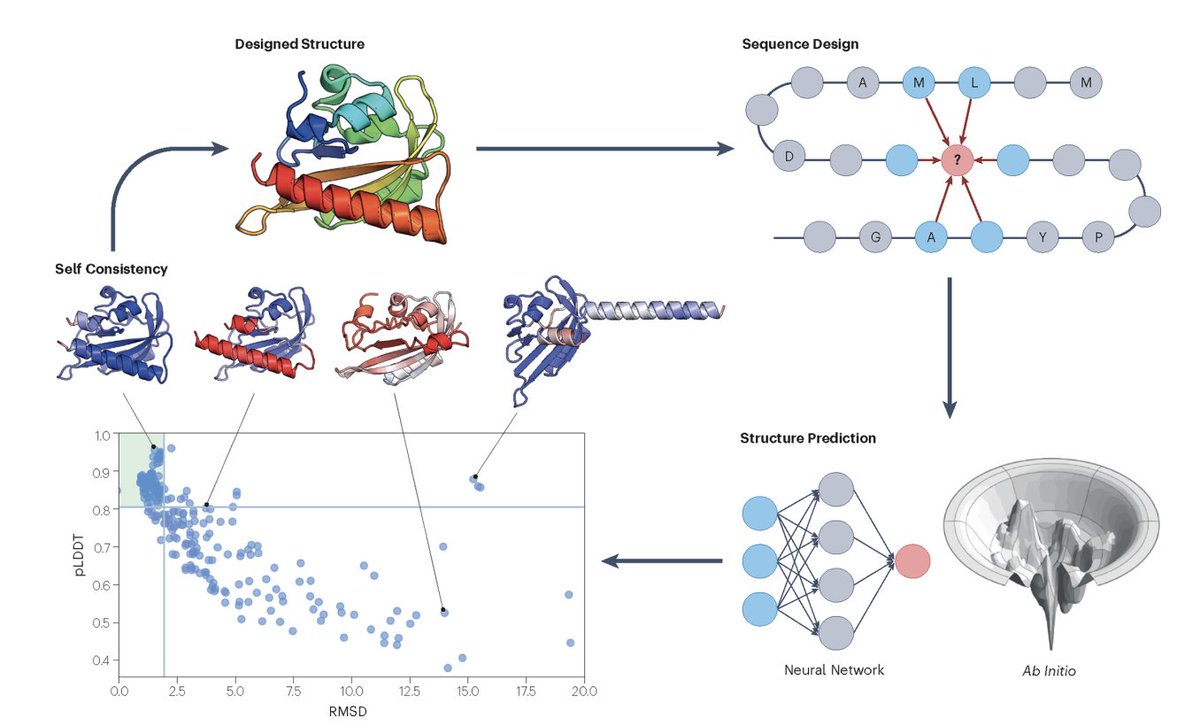
8. RedNet: a multiscale graph transformer for fixed-backbone binder design that encodes both backbone geometry and side-chain/atom-level context, then autoregressively decodes sequences. It is evaluated not only by sequence recovery/likelihood but also by downstream binding-related proxies.
9. A key design contribution is contrastive decoding/scoring for specificity: sequences are generated to increase on-target compatibility while simultaneously decreasing off-target compatibility, enabling discrimination between highly structurally similar targets; evaluations include zero-shot binding affinity correlation on SKEMPI and selectivity tests via Rosetta energetics and AlphaFold3 cofolding-based criteria.
10. Overall takeaway: the thesis argues that combining (a) structure-native representations (graphs with local frames, multiscale atom/residue/surface context), (b) PLM-derived evolutionary/attention signals, and (c) principled search/decoding (pairing interologs; contrastive decoding) yields complementary gains for both predicting complexes and designing selective binders.
💻Code: github.com/zw2x/glinter
📜Paper: arxiv.org/abs/2605.11189
#ComputationalBiology #ProteinDesign #ProteinStructure #ProteinComplexes #DeepLearning #GraphNeuralNetworks #ProteinLanguageModels #AlphaFold #Bioinformatics
2
20
111
5,146

May 14
Excited to share that ProtoMech, our mechanistic interpretability tool for protein language models, has been accepted to ICML 2026! 🎉
#ICML #ICML2026 #MechanisticInterpretability #ProteinLanguageModels #AI4Science #ComputationalBiology #MachineLearning #Interpretability #BioAI #DeepLearning
May 14

Can we learn how protein language models (pLMs) work?👀
As a matter of fact, yes! I'm excited to shared that our paper, "Protein Circuit Tracing via Cross-layer Transcoders", has been accepted into ICML 2026! Check out our findings at protmech.github.io/ (1/n)
2
2
12
1,182
Proteinopd: Towards effective and efficient preference alignment for protein design
1 ProteinOPD is a multi-objective preference alignment framework for protein language models (PLMs) that improves target protein properties while preserving “designability” (naturalness/structural plausibility), addressing a common failure mode of post-training alignment: catastrophic forgetting.
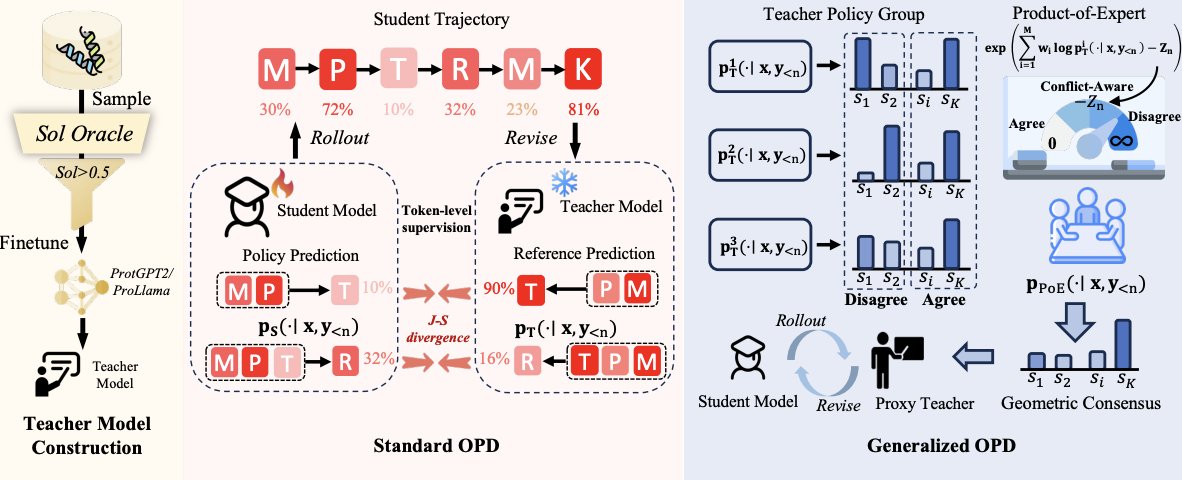
2 The key idea is to adapt a pretrained PLM into several preference-specific teacher models (one per property) using lightweight oracle-guided SFT on a few hundred high-scoring sequences, then distill these teachers into a single shared student using token-level On-Policy Distillation (OPD) on the student’s own rollouts.
3 Why OPD for protein alignment: unlike mode-covering SFT, OPD is mode-seeking and provides dense token-level supervision on the student-visited states, which helps reduce exposure bias and better retains pretrained generative competence compared with directly training on curated “good” sequences.
4 ProteinOPD’s main methodological contribution is extending OPD to multi-objective alignment via a generalized multi-teacher target: at each generation step, it constructs a geometric consensus distribution as a normalized product-of-experts (PoE) over teacher next-token distributions (weighted geometric mean).
5 This PoE consensus is derived as the solution to minimizing the weighted sum of KL divergences from a target distribution q to each teacher, yielding a principled “consensus over coverage” target that emphasizes tokens jointly supported across objectives rather than tokens favored by any single objective.
6 A stability insight: the PoE normalization term Zn is always ≤ 0, and −Zn serves as a free, token-level disagreement/conflict measure among teachers; when objectives conflict strongly, the consensus signal is bounded, improving training stability under antagonistic or noisy preferences.
7 Experimental setup spans unconditional generation (ProtGPT2) and conditional family-guided generation (ProLLaMA on a lysozyme-like superfamily). Designability is evaluated with PPL, novelty vs UniProtKB/ training sets, and ProTrek (conditional semantic consistency). Preference alignment uses oracles: ESMFold-derived pLDDT/PAE (foldability), Protein-Sol (solubility), and TemBERTure (thermostability). Overall trade-offs are summarized by hypervolume (HV).
8 Multi-objective unconditional results: ProteinOPD achieves the best overall trade-off (HV 0.62), outperforming MoMPNN (0.46) and ProGen2 (0.44). Relative to ProtGPT2, it improves pLDDT ( 14.8%), solubility ( 16.9%), and thermostability ( 54.2%), while also improving designability proxies (PPL reduced by 83.7%) and increasing novelty vs training data ( 9.4%).
9 Single-objective analysis shows OPD can transfer property gains from SFT teachers while mitigating SFT-induced novelty/designability loss; in conditional generation it also improves condition fidelity (ProTrek 10% vs Teacher-SFT), consistent with OPD correcting the student on its own visited trajectories rather than forcing coverage of a narrow curated dataset.
10 Efficiency: compared with RL-based alignment (e.g., ProtRL/GRPO-style), ProteinOPD reaches comparable alignment quality with ~8× less training time, benefiting from dense token-level distillation rather than sparse sequence-level rewards and repeated oracle rollouts; teacher construction remains data-efficient (good performance with ~100 filtered sequences, improved further with stricter oracle thresholds).
💻Code: github.com/THU-AI4S/ProteinO…
📜Paper: arxiv.org/abs/2605.10189
#ProteinDesign #ProteinLanguageModels #Alignment #PreferenceLearning #Distillation #MultiObjectiveOptimization #ComputationalBiology #SyntheticBiology #MachineLearning
1
4
29
2,249
ProtSpace: Protein Universe in Your Browser
1 ProtSpace is a web-native viewer for protein language model (pLM) embedding spaces that runs entirely client-side, enabling interactive exploration of up to ~573K Swiss-Prot proteins in the browser with no installation, no login, and no data upload (privacy by design).
2 The core idea is to move beyond sequence-similarity networks (e.g., BLAST-derived graphs) and instead visualize learned embedding neighborhoods that can capture functional/structural relationships even when sequence similarity is weak, using interactive 2D projections as a hypothesis-generation interface.
3 A key engineering contribution is scale: WebGL-accelerated rendering plus a quadtree spatial index yields responsive hover/click/selection at hundreds-of-thousands of points; pan/zoom stays ~constant-time, while annotation switching and selection scale roughly linearly with dataset size in their benchmark suite.
4 The pipeline is designed for “bring your own proteins”: users can start from FASTA, a UniProt query, or precomputed HDF5 embeddings. Preparation is available via a zero-install Google Colab notebook or a Python CLI (protspace prepare), with caching and a YAML run log for reproducibility.
5 ProtSpace supports 12 embedding models via the Biocentral API (including ProtT5, multiple ESM-2 variants, Ankh, and ESM-C). For known proteins, UniProt’s precomputed ProtT5 embeddings can eliminate the need for local GPUs.
6 The annotation system is unusually rich for an embedding viewer: 38 switchable annotation types are automatically retrieved from five sources (UniProt, InterPro, NCBI Taxonomy, TED structural domains via AlphaFold DB, and Biocentral predictors), and can be augmented/overridden by user-provided CSV metadata.
7 It explicitly represents uncertainty/provenance: UniProt-derived evidence codes (ECO) are preserved and shown for key fields (e.g., GO aspects, EC numbers, subcellular localization, protein families), while InterPro domain hits expose per-domain scores and TED domains include pLDDT-based confidence.
8 A distinctive visualization feature is multi-label rendering as per-point pie charts (e.g., domain architectures such as multiple Pfam hits), allowing users to see compositional patterns across embedding neighborhoods rather than forcing single-label coloring.
9 ProtSpace integrates structure into the same exploratory loop: selecting a protein can open an embedded Mol* viewer that loads AlphaFold predictions via 3D-Beacons, enabling rapid cross-checks between embedding proximity, annotations, and structural context.
10 The paper’s examples emphasize multi-scale biological use: (i) Swiss-Prot-wide organization separates broad domains of life; (ii) joint human–Drosophila projections reveal overlap for conserved families and isolated regions enriched for lineage-specific families; (iii) beta-lactamase superfamily maps recover known Ambler-class structure and flag candidates for misannotation (e.g., proteins clustering with mechanistically different groups) and potential novel family clusters.
💻Code: github.com/tsenoner/protspac…
📜Paper: biorxiv.org/content/10.64898…
#Bioinformatics #ComputationalBiology #ProteinLanguageModels #Embeddings #ProteinFunction #UniProt #InterPro #AlphaFold #WebGL #Visualization

8
39
2,608
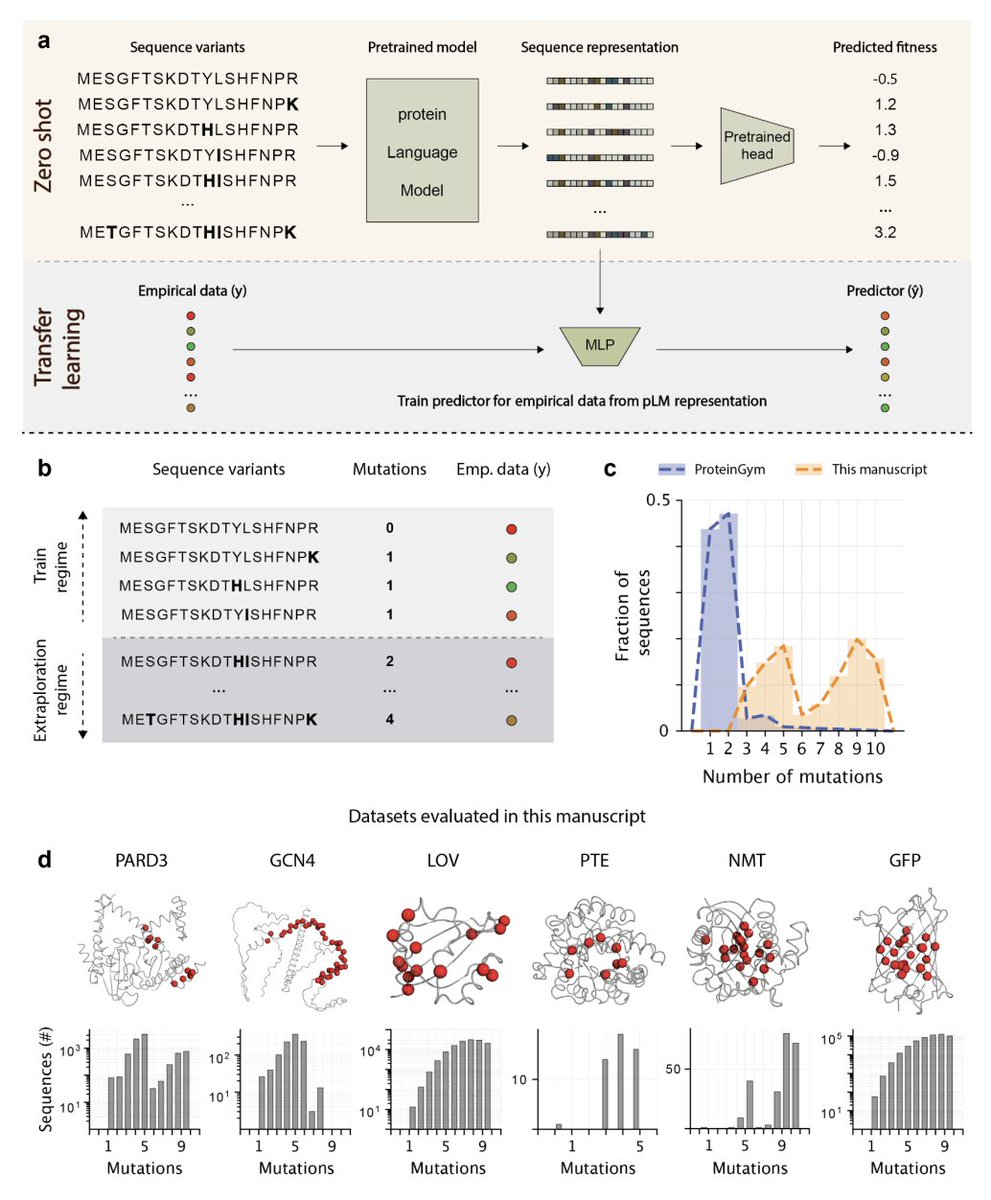
Simple Baselines Rival Protein Language Models in Mutation-Dense Design Tasks
1. The study benchmarks popular protein language models (pLMs) specifically in mutation-dense, high-order combinatorial design landscapes, where the goal is to extrapolate from low-order measured mutants to unseen higher-order combinations (a design-relevant OOD setting often missing from standard benchmarks).
2. Across six dense multi-mutant datasets (FuncLib/htFuncLib: GFP, PTE, NMT; ProteinGym: LOV, PARD3, GCN4), zero-shot pLM scoring showed weak separation of functional vs non-functional variants (often near-chance ROC AUC ~0.5–0.6) and modest phenotype correlations (Spearman ρ ranging roughly from negative to moderate positive values).
3. A key baseline result: zero-shot pLM scores were statistically indistinguishable from a simple homology-based evolutionary prior (PSSM). In other words, for ranking/triage in these combinatorial active-site regimes, pLMs did not consistently outperform “classical” sequence statistics computed from an MSA.
4. For transfer learning, the authors introduce an extrapolation-controlled protocol: train on variants up to mutational order k_train and test on a fixed higher order k_test, explicitly quantifying performance degradation with increasing train–test mutational gap (rather than using only random splits).
5. In transfer learning across eight pLMs (ESM2 sizes from 8M to 3B, ProtBert, ProGen2 variants), model identity/scale/architecture contributed little: pLMs were largely indistinguishable from each other, and in ~96% of pLM-to-pLM comparisons differences were not statistically significant.
6. The dominant driver of predictive performance was the train–test mutation gap (how far the test variants are from what was measured), not parameter count or training cost. Notably, ESM2-8M could match or exceed ESM2-3B in these extrapolative regimes despite ~1000x fewer parameters.
7. A second central baseline: in supervised prediction, a simple one-hot encoding (OHE) of substitutions paired with the same downstream MLP rivaled—and often matched or outperformed—the best pLM representation, both under controlled extrapolation and under standard random train/test splits across training set sizes.
8. Beyond prediction, the paper tests model-guided landscape exploration: ranking beneficial vs deleterious single mutations and nominating combinatorial “neighborhoods” to define screening libraries enriched for gain-of-function variants. pLMs again did not significantly outperform PSSMs on mutation ranking or library enrichment metrics.
9. The authors’ interpretation: pretraining on natural sequences may encode “tolerated” evolutionary variation but is not a reliable standalone signal for gain-of-function combinatorial design, especially under strong epistasis typical of active sites. They argue pLMs may need explicit biophysical/structural priors or integration with structure-based approaches to improve design utility.
10. Practical contribution: the mutation-dense datasets used here are deposited to ProteinGym to support future benchmarking/training, and the paper emphasizes that any new “general-purpose” design claim should be tested against strong, simple baselines (PSSM, OHE) under extrapolation-controlled splits.
💻Code: github.com/drprfitay/fitness…
📜Paper: biorxiv.org/content/10.64898…
#ProteinDesign #ProteinLanguageModels #MachineLearning #ComputationalBiology #Benchmarks #Epistasis #DirectedEvolution #Bioinformatics #Rosetta #ProteinEngineering
3
14
46
3,938
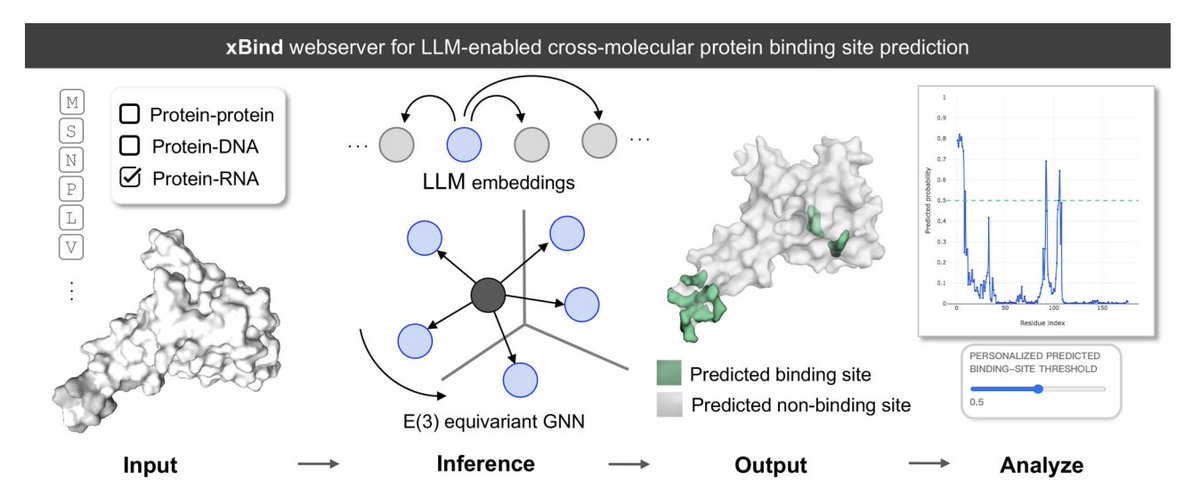
xBind: An Integrated Webserver for Large Language Model-Enabled Cross-Molecular Protein Binding Site Prediction
1. xBind is a unified webserver for residue-level binding-site prediction across three interaction types: protein–protein, protein–DNA, and protein–RNA, aiming to remove the usual “one tool per modality” fragmentation.
2. The core modeling idea is to combine ESM-2 protein language model embeddings with sequence- and structure-derived features, then learn on 3D graphs using symmetry-aware E(3)-equivariant GNNs to better respect molecular geometry.
3. A practical highlight is that xBind accepts either (a) a single-chain protein sequence (FASTA) or (b) a monomer structure (PDB/mmCIF). For sequence-only inputs, it can run AlphaFold2 on the fly to produce a structure for structure-aware inference.
4. Under the hood, xBind exposes two independently trained frameworks: EquiPPIS for protein–protein interfaces and EquiPNAS for protein–DNA/RNA interfaces, both designed to be robust to imperfect structures (including AlphaFold models).
5. Benchmarking emphasizes performance on AlphaFold2-predicted structures, reflecting real-world usage where experimental complexes are missing. Reported comparisons show statistically significant gains (95% confidence, P < 0.05) over strong baselines using ROC-AUC and PR-AUC across multiple test sets.
6. Example performance (AlphaFold inputs): for protein–protein, EquiPPIS improves over GraphPPIS on Test_60 (ROC-AUC 0.7912 vs 0.7667; PR-AUC 0.4516 vs 0.3956). For protein–DNA, EquiPNAS leads on Test_181 (ROC-AUC 0.9159; PR-AUC 0.3717) and Test_129 (ROC-AUC 0.9387; PR-AUC 0.569). For protein–RNA, EquiPNAS improves over GraphBind on Test_117 (ROC-AUC 0.8856 vs 0.7942; PR-AUC 0.3118 vs 0.2019).
7. The web UI is built for interpretability and calibration: it provides interactive sequence views, 3D visualization of predicted interface residues, per-residue likelihood plots, and user-adjustable probability thresholds to tune precision/recall for different downstream needs.
8. Output artifacts are designed for reuse: downloadable structure used for prediction, residue-level probability files, and a packaged archive with complete outputs and run metadata; jobs can be public or private, with optional email notification.
9. The paper also outlines near-term directions: calibrating predictions using residue-wise uncertainty signals (e.g., experimental B-factors or AlphaFold pLDDT) and incorporating conformational dynamics/multistate behavior to better model binding under structural heterogeneity.
📜Paper: doi.org/10.1093/nar/gkag425
#Bioinformatics #ComputationalBiology #ProteinScience #StructuralBioinformatics #DeepLearning #GraphNeuralNetworks #ProteinLanguageModels #ESM2 #AlphaFold #ProteinInteractions #NARWebServerIssue
3
1,336
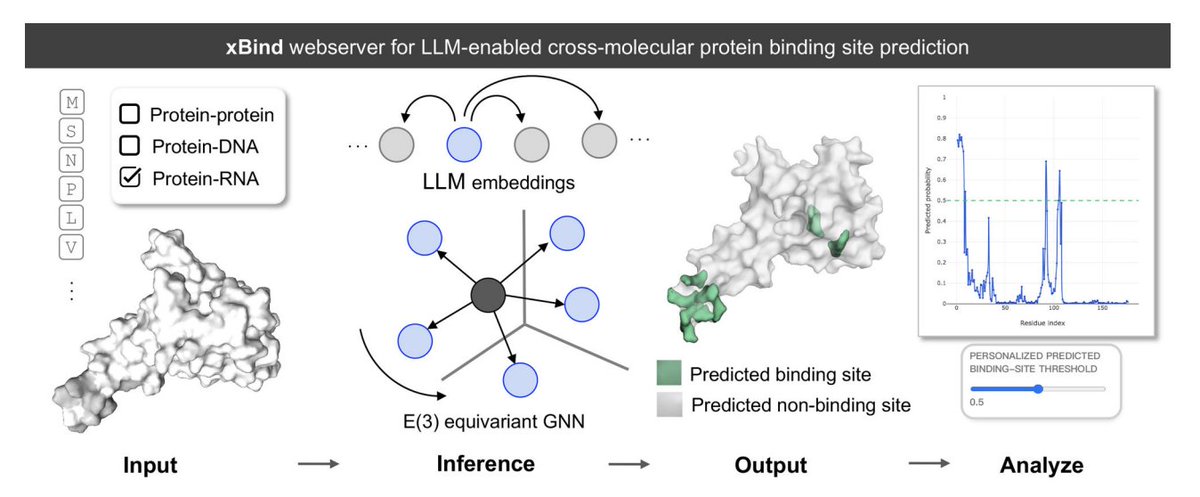
xBind: An Integrated Webserver for Large Language Model-Enabled Cross-Molecular Protein Binding Site Prediction
1. xBind is a unified webserver for residue-level binding-site prediction across three interaction types: protein–protein, protein–DNA, and protein–RNA, aiming to remove the usual “one tool per modality” fragmentation.
2. The core modeling idea is to combine ESM-2 protein language model embeddings with sequence- and structure-derived features, then learn on 3D graphs using symmetry-aware E(3)-equivariant GNNs to better respect molecular geometry.
3. A practical highlight is that xBind accepts either (a) a single-chain protein sequence (FASTA) or (b) a monomer structure (PDB/mmCIF). For sequence-only inputs, it can run AlphaFold2 on the fly to produce a structure for structure-aware inference.
4. Under the hood, xBind exposes two independently trained frameworks: EquiPPIS for protein–protein interfaces and EquiPNAS for protein–DNA/RNA interfaces, both designed to be robust to imperfect structures (including AlphaFold models).
5. Benchmarking emphasizes performance on AlphaFold2-predicted structures, reflecting real-world usage where experimental complexes are missing. Reported comparisons show statistically significant gains (95% confidence, P < 0.05) over strong baselines using ROC-AUC and PR-AUC across multiple test sets.
6. Example performance (AlphaFold inputs): for protein–protein, EquiPPIS improves over GraphPPIS on Test_60 (ROC-AUC 0.7912 vs 0.7667; PR-AUC 0.4516 vs 0.3956). For protein–DNA, EquiPNAS leads on Test_181 (ROC-AUC 0.9159; PR-AUC 0.3717) and Test_129 (ROC-AUC 0.9387; PR-AUC 0.569). For protein–RNA, EquiPNAS improves over GraphBind on Test_117 (ROC-AUC 0.8856 vs 0.7942; PR-AUC 0.3118 vs 0.2019).
7. The web UI is built for interpretability and calibration: it provides interactive sequence views, 3D visualization of predicted interface residues, per-residue likelihood plots, and user-adjustable probability thresholds to tune precision/recall for different downstream needs.
8. Output artifacts are designed for reuse: downloadable structure used for prediction, residue-level probability files, and a packaged archive with complete outputs and run metadata; jobs can be public or private, with optional email notification.
9. The paper also outlines near-term directions: calibrating predictions using residue-wise uncertainty signals (e.g., experimental B-factors or AlphaFold pLDDT) and incorporating conformational dynamics/multistate behavior to better model binding under structural heterogeneity.
📜Paper: doi.org/10.1093/nar/gkag425
#Bioinformatics #ComputationalBiology #ProteinScience #StructuralBioinformatics #DeepLearning #GraphNeuralNetworks #ProteinLanguageModels #ESM2 #AlphaFold #ProteinInteractions #NARWebServerIssue
1
3
37
2,274