
EasyNano: rapid epitope-targeted nanobody CDR design via differentiable distogram optimization with ESMFold2
1 EasyNano is a rapid pipeline for epitope-targeted nanobody CDR redesign that runs in ~10–20 minutes per target on a high-end personal workstation, aiming to make “design-to-candidate” iteration practical without GPU clusters.
2 The core idea is to optimize CDR residue logits by gradient descent through the ESMFold2-Fast distogram (a differentiable proxy), rather than trying to directly optimize ipTM (expensive and not practical as an inner-loop objective).
3 EasyNano introduces an explicit epitope-targeting objective: a dedicated CDR-to-epitope proximity loss (ELU penalty when expected CDR→epitope distance exceeds 8 Å), enabling user-specified epitope steering instead of “bind anywhere”.
4 To prevent framework pose drift during optimization, EasyNano computes a structure prior using full ESMFold2 (1.3B) on the WT framework–target complex, then constrains optimization with a CA-coordinate distogram mask prior; this anchoring is critical for stable epitope-focused design.
5 The method uses a three-stage workflow: (i) full-model structure prior (~30 s), (ii) differentiable CDR optimization with ESMFold2-Fast (~10–17 min; 60 steps; Adam; cosine temperature schedule), (iii) full ESMFold2 evaluation (~15 s per candidate) to obtain calibrated ipTM/pTM for ranking.
6 A practical insight from systematic sweeps: the wild-type logit initialization bias (β) is the key hyperparameter controlling CDR mutability. Too high (β≥5) freezes CDRs; too low (β≤1) causes chaotic drift. β≈2 (with moderate prior weight) enables meaningful, stable mutation.
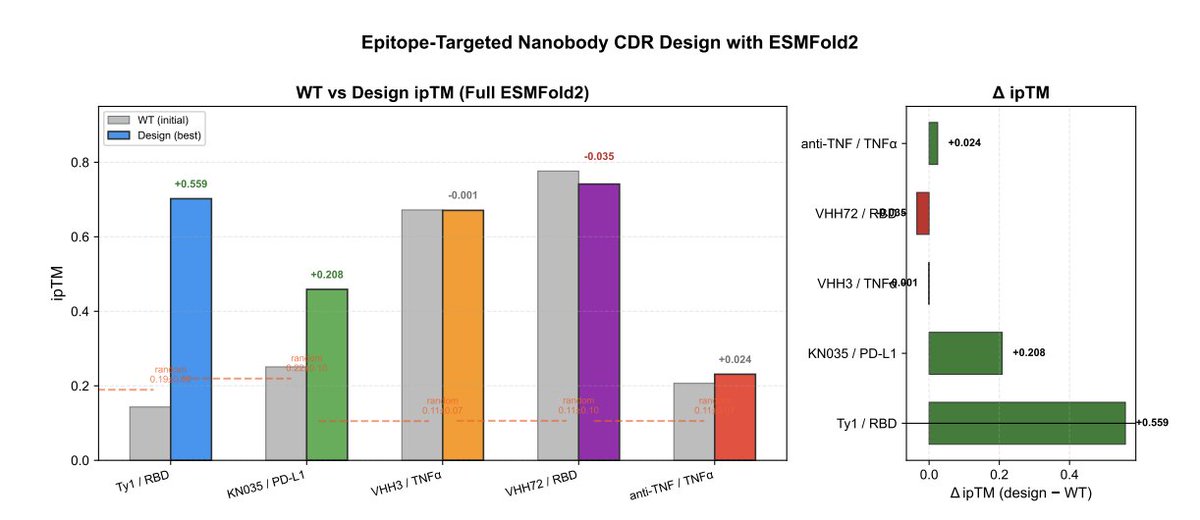
7 On weak binders, EasyNano can yield large ipTM gains: Ty1/RBD improved from 0.143 to 0.702 ( 0.559; 5.7σ above random CDR baseline), with 11/22 CDR mutations and reduced CDR→epitope distance (16.6 Å → 10.7 Å).
8 It also improves clinically relevant cases while respecting constraints: KN035/PD-L1 increased ipTM 0.251 → 0.459 ( 0.208; 2.2σ), introducing 7/32 mutations while preserving the H3 disulfide, consistent with constrained-but-targeted optimization.
9 On already-strong binders (e.g., VHH72/RBD and VHH3/TNFα), EasyNano largely preserves ipTM (small ∆), suggesting the approach does not necessarily degrade optimized interfaces when headroom is limited.
10 De novo scenario: starting from a manually docked non-cognate framework near AQP4 loop C, CDR-only design improved ipTM 0.117 → 0.538 (4.6-fold). Multi-seed runs revealed distinct local minima; a single framework micro-tuning mutation (W116Y) stabilized the high-ipTM basin, highlighting a practical interplay between pose basins and CDR optimization.
💻Code: github.com/[organization]/Ea…
📜Paper: arxiv.org/abs/2606.12772
#Nanobody #ProteinDesign #AntibodyEngineering #ComputationalBiology #ESMFold #DeepLearning #DifferentiableOptimization #EpitopeTargeting #Bioinformatics
6
37
2,038

3/ Along the way: the malaria bromodomain PfBDP1, the combinatorial logic of the ATAD2/B readers, and a new push into AI-driven protein design with NIH & NSF support and a phenomenal group of trainees. Here’s to the next 5. 🎉 #epigenetics #proteindesign
1
49
SurfDesign: Effective Protein Design on Molecular Surfaces
1. SurfDesign reframes protein design around molecular surfaces (shape physicochemical complementarity), aiming to better control functional regions like binding interfaces and enzyme pockets compared with backbone-only conditioning.
2. Core technical idea: treat molecular surfaces as continuous geometric manifolds rather than unordered point clouds/meshes, so the model can leverage local tangent structure, curvature, and directional consistency that are important for interaction-centric design.
3. The method builds an oriented surface point cloud Q where each point includes coordinates, a unit normal vector, and physicochemical attributes (e.g., hydrophobicity, charge, H-bond features). Surfaces are generated via PyMOL/MSMS and denoised with Gaussian smoothing; no residue identities, MSAs, or functional labels are used in surface generation.
4. SurfDesign introduces a Surface-conditioned Equivariant Message Passing (SEMP) encoder: SE(3)-equivariant updates use invariant radial distances, curvature descriptors (from local covariance eigenvalues), and directional angular features derived from surface normals (two intersection angles one dihedral angle).
5. Directionality is encoded with spherical Fourier–Bessel bases over distances and angles; messages are attention-reweighted and used to update both node features and coordinates, with optional per-layer recomputation of normals/curvatures to stay consistent as coordinates evolve.
6. To address limited surface-structure paired data and improve sequence priors, SurfDesign integrates pretrained protein language models (PLMs) via parameter-efficient fine-tuning (hybrid PEFT: structural adapter LoRA), trained with conditional masked language modeling rather than autoregressive decoding.
7. Binder design benchmark (6 targets) uses AF2 pAE_interaction as a functional proxy. SurfDesign achieves the best average pAE_interaction (15.85) and the highest overall success rate (30.14%), outperforming SurfPro (surface-conditioned baseline) and backbone-only baselines like ProteinMPNN, PiFold, and LM-DESIGN.
8. Enzyme design benchmark (5 enzyme–substrate systems; leakage-controlled by excluding overlaps with CATH pretraining) uses ESP score as a proxy for enzyme–substrate compatibility. SurfDesign attains the best average success rate (47.30%) and the best average ESP under greedy decoding (0.9058), with gains persisting in a zero-shot substrate setting.
9. Inverse folding is positioned as a diagnostic for structural compatibility (not “recovering the native sequence”). SurfDesign reports strong results on CATH splits, including perplexity 2.41 and AAR 74.13% on CATH 4.2, plus improved surface recovery metrics (IoU/CD/NC) versus PLM-based baselines; scaling larger PLMs further improves recovery.
💻Code: github.com/smiles724/SurfDes…
📜Paper: arxiv.org/abs/2606.07567
#ProteinDesign #ComputationalBiology #GeometricDeepLearning #ProteinEngineering #EnzymeDesign #ProteinBinding #EquivariantNetworks #ProteinLanguageModels #KDD2026
15
74
3,913
Towards Coevolution-Aware Ancestral Sequence Reconstruction
1. The paper proposes a practical way to make ancestral sequence reconstruction (ASR) “epistasis-aware”: it keeps the standard phylogenetic posterior uncertainty per site, but enforces residue–residue constraints learned from extant sequences via Direct Coupling Analysis (DCA).
2. Core motivation: common ASR pipelines assume sites evolve independently. This can (a) over-concentrate probability into a single, over-idealized MAP ancestor, or (b) make posterior sampling produce implausible, often non-functional sequences because it ignores coevolutionary compatibility across sites.
3. Method in three steps: (i) run a fast site-independent Bayesian ASR (Yang-style pruning) to obtain per-site posteriors at the root; (ii) sample M=1000 candidate root sequences from the factorized posterior; (iii) “reshuffle” candidates by swapping residues at the same site between sequences, accepting swaps by a Metropolis criterion based on the change in DCA energy.
4. Key design choice: swaps are column-wise, so single-site amino-acid frequencies (the site-independent marginals/posteriors) are exactly preserved, while pairwise consistency is improved by lowering DCA energy. This yields an ensemble that is simultaneously phylogenetically consistent (marginals) and coevolution-compatible (pairwise couplings).
5. A major contribution is the benchmarking setup: the authors build a controlled forward-evolution simulator driven by a DCA model, evolving sequences along a real inferred tree topology. This provides ground-truth internal/root sequences under realistic epistatic constraints—something standard site-independent simulators cannot represent.
6. They explore evolutionary regimes by globally scaling branch lengths (µgen), spanning shallow to deep divergence, and they vary the “mutability” of the true root using a DCA-derived metric (Context-Dependent Entropy, CDE). Result: ASR difficulty depends strongly on both divergence time and ancestral mutability/epistatic constraint strength.
7. In β-lactamases (and similarly in a DNA-binding domain family in the SI), coevolution-aware reshuffled ensembles improve reconstruction when ancestral states are strongly epistatically constrained. Compared to naive posterior sampling, reshuffling drives candidates toward DCA energies typical of natural families while maintaining phylogenetic marginals.
8. The analysis highlights a subtle bias: MAP (and even leaf consensus) can become “over-optimized” in DCA energy relative to the true ancestor, potentially explaining why resurrected MAP ancestors are often reported as unusually stable or promiscuous—an effect consistent with model-driven over-idealization rather than true history.
9. For selecting candidates without knowing the ground truth, they rank sequences by site-independent posterior probability PR and examine the top N candidates. Coevolution-aware top candidates tend to match or sometimes beat MAP in Hamming distance to ground truth, while improving statistical plausibility (lower DCA energy) and structural plausibility by ESMFold metrics (higher pLDDT; lower RMSD to a GT reference structure in many settings).
10. Limitations and outlook: the method currently reconstructs nodes independently (not a coherent joint sampling of trajectories across the whole tree). Still, it offers a tractable bridge between single-sequence MAP ASR and unconstrained posterior sampling, explicitly injecting coevolutionary constraints in an interpretable way.
📜Paper: biorxiv.org/content/10.64898…
#AncestralSequenceReconstruction #ASR #Coevolution #Epistasis #DCA #ProteinEvolution #Phylogenetics #ComputationalBiology #ProteinDesign #Bioinformatics

3
24
2,095
Discriminator-guided Inverse Folding for Multi-property Protein Design
1 DGIF introduces a plug-and-play way to steer an inverse-folding model toward multiple protein properties at once, without fine-tuning the generative model and without needing multi-property-labeled datasets.
2 Core idea: during autoregressive sequence generation, DGIF backpropagates gradients from an auxiliary discriminator into the decoder’s internal history state (KV-cache / “history states”), then re-samples the next residue from the updated distribution—repeating this at every step.
3 The discriminator is a composition of multiple single-property predictors. Each predictor can be trained independently on a dataset labeled for only that property, and DGIF combines their signals with weights (beta_i) to perform multi-objective optimization.
4 DGIF is implemented on top of ESM-IF1, producing three variants: DG-Thermo (thermostability), DG-Sol (solubility), and DG-Dual (thermostability solubility). The base inverse-folding model parameters remain unchanged.
5 For thermostability guidance, the paper trains a ΔΔG predictor using ESM-IF1 representations on the Megascale dataset (≈700k mutation–stability pairs), with additional evaluation on FireProt and S669. The predictor outperforms several classic baselines (e.g., FoldX/Rosetta/Thermonet) and is competitive with ThermoMPNN.
6 DG-Thermo improves design outcomes vs unguided ESM-IF1 on: (i) average top-K recall for stabilizing mutations on Megascale test proteins, and (ii) “success rate” of full-sequence designs that both improve predicted stability (ΔΔG > 1.0 kcal/mol) and maintain foldability (predicted structure RMSD < 2 Å).
7 Mechanistic signals emerge naturally: DG-Thermo-designed proteins show more salt bridges and hydrophobic interactions, and amino-acid composition shifts consistent with thermophilic trends (e.g., increased L/P/R/W and decreased D/K/M/Q), despite these rules not being explicitly encoded as constraints.
8 MD validation: for xylanase at 450 K (100 ns), DG-Thermo variants maintain structure (lower RMSD, higher secondary-structure retention) compared with wild type and an unguided ESM-IF1 design; additional CATH-sampled scaffolds show similar stability gains in MD.
9 Solubility guidance: a binary solubility predictor (ESM-IF1 representations MLP) is trained on Khurana et al. and tested on Chang et al.; DG-Sol improves top-K recall on SoluProtMutDB and increases design success rates under joint criteria (better predicted solubility RMSD < 2 Å). On membrane proteins, DG-Sol designs increase surface polar residue proportion, consistent with higher solubility.
10 Multi-property optimization: DG-Dual jointly applies thermostability and solubility predictors and shifts designs toward the Pareto front (better stability/solubility trade-offs) on CATH redesign tasks. Wet-lab validation on Rhodococcus ruber alcohol dehydrogenase (RrADH) tests 10 DG-Dual-suggested single mutations: all improve solubility; 8/10 increase melting temperature. Examples include A50E (≈2x ELISA solubility signal 2.79 °C Tm) and S223A ( 6.47 °C Tm with concurrent solubility gain).
💻Code: github.com/aweqardf/ESM-IF1-…
📜Paper: doi.org/10.1002/advs.75988
#ProteinDesign #InverseFolding #MultiObjectiveOptimization #ESM #ComputationalBiology #MachineLearning #Thermostability #Solubility #ProteinEngineering

5
23
1,430
Flexible Kernels for Protein Property Prediction
1. The paper introduces LOCK-GP: Gaussian processes with a new protein sequence kernel that combines evolutionary substitution matrices (e.g., BLOSUM) with an explicit “local linearity” inductive bias to model protein property landscapes from sparse experimental data.
2. Key kernel idea (LOCK: Locally Linear Correlation Kernel): replace one-hot “same/different” comparisons with amino-acid similarity from substitution matrices, and learn landscape-specific Hadamard-power exponents to tune how strongly similarities are amplified/attenuated while preserving kernel validity.
3. A central technical observation: many BLOSUM matrices are not only PSD but also infinitely divisible, so elementwise exponentiation by any positive power preserves PSD. This enables learnable exponents inside the GP kernel without breaking positive semidefiniteness.
4. LOCK is built from (i) an additive “linear” correlation kernel and (ii) a multiplicative “RBF-like” correlation kernel, then combined so predictions are nuanced and non-linear near training data but revert to a robust linear predictor farther away (avoiding both aggressive linear extrapolation and mean-reversion to the prior mean).
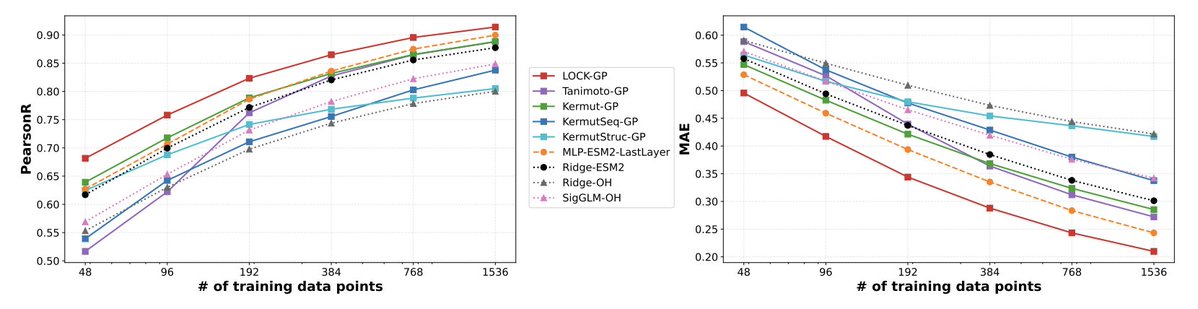
5. Benchmarking is extensive: 30 predictors evaluated across 21 protein property datasets (thermostability, binding affinity, fluorescence, capsid viability, etc.) under three regimes: i.i.d. CV, Hamming-distance extrapolation, and an “unseen mutations” OOD regime where test sequences include mutations absent from training.
6. Results highlight data efficiency and uncertainty quality: across datasets and training sizes (e.g., 48–1536 points), LOCK-GP is typically best or near-best on correlation and error metrics, and shows strong calibration via proper scoring rules like CRPS; uncertainty improves notably when local linearity is included.
7. A notable empirical takeaway: a sequence-only LOCK-GP that relies on a small substitution-matrix prior can frequently outperform or match baselines that depend on large foundation models (e.g., ESM-2 embeddings, structure features, ProteinMPNN-derived features), especially in extrapolation and OOD “unseen mutation” settings where high-dimensional embeddings can be fragile.
8. The paper generalizes LOCK to CLOCK (structure-conditioned LOCK): positional structure embeddings from a foundation model are mapped to position-specific amino-acid correlation matrices (parameterized as exp(-||z_a - z_a'||^2)), effectively learning structure-aware substitution behavior that can be used “zero-shot” as a kernel prior and then refined by GP training.
9. Multi-task learning: CLOCK-GP is trained across 371 thermostability landscapes (Tsuboyama et al.), showing that learning a shared, structure-conditioned kernel across landscapes yields strong performance; CLOCK-GP is especially competitive in low-landscape regimes (e.g., training on 10 landscapes), and learned correlations are interpretable (e.g., proline preferences near helix N-termini; arginine favored on surfaces vs cores).
10. Additional demonstrations: LOCK-GP supports GP-based Bayesian optimization via Thompson sampling to control exploration/diversity in design, and extends to binary classification (e.g., quantized fluorescence) with strong accuracy scaling with dataset size.
💻Code: github.com/generatebio/lock_…
📜Paper: arxiv.org/abs/2606.11057
#ComputationalBiology #ProteinEngineering #GaussianProcesses #MachineLearning #Kernels #ProteinDesign #UncertaintyQuantification #MultiTaskLearning #FoundationModels #Bioinformatics
2
11
1,110
Flexible Flows for Biological Sequence Design
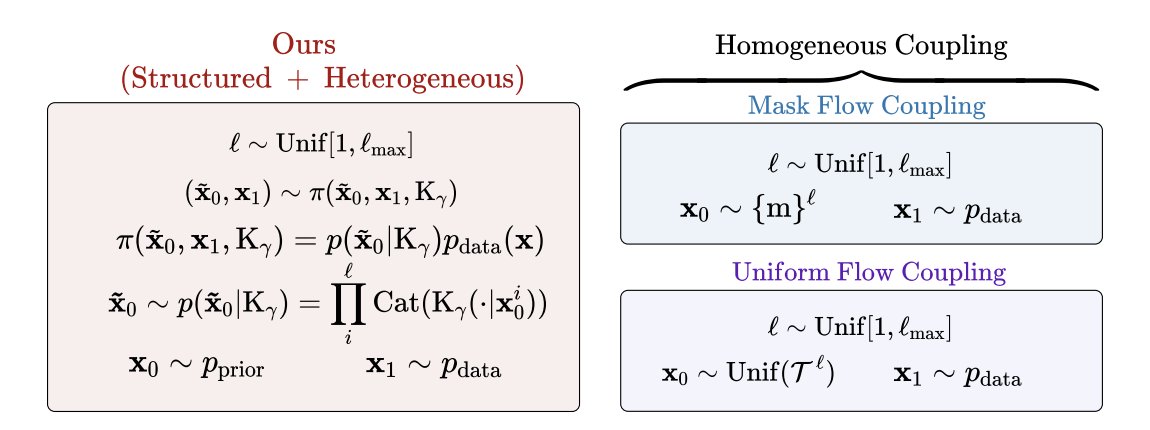
1. FlexFlow reframes discrete flow matching for biological sequences by changing the coupling (forward endpoint pairing) rather than the training objective: a structured, biology-informed coupling uses substitution matrices (e.g., BLOSUM for proteins; JC69/HKY85-style biases for nucleotides) to tilt the source distribution toward evolutionarily plausible neighborhoods.
2. The key idea is to keep the standard token-wise mixture path and CTMC machinery intact, but swap the usual “uninformative” couplings (uniform/masked) with a transition kernel Kγ that encodes preferred substitutions; when Kγ is uniform, the method reduces to the standard uniform coupling.
3. For variable-length generation, FlexFlow builds on Edit Flows by parameterizing reverse-time CTMC rates via edit operations (insertion, deletion, substitution). Instead of treating positions independently, it introduces a shared global latent r that conditions per-position edit decisions, coupling token-level operations through sequence-level context.
4. FlexFlow adds test-time control over edit behavior: operation probabilities are temperature-scaled and modeled with a Dirichlet prior over {ins, sub, del}. By changing Dirichlet concentrations α post-hoc, users can bias generation toward more insertions vs substitutions vs deletions without retraining, effectively acting as an “operation budget controller.”
5. The paper proposes latent classifier-free guidance (CFG) as an alternative to rate-space guidance: it performs CFG by interpolating conditional/unconditional latents (rc and r∅) in continuous space, then uses the guided latent to drive all edit operations jointly—aiming for more globally coherent conditioning than token-wise rate guidance.
6. The latent guidance has a probabilistic interpretation: under Gaussian conditional/unconditional latent encodings and sufficiency assumptions, the guidance direction corresponds to the score of an implicit classifier p(c|r), making the latent interpolation analogous to a gradient ascent step on log p(c|r).
7. Training uses an augmented alignment space with a blank token ε to make edit-based objectives tractable: alignments define edit sequences between endpoints, and a Bregman-divergence-style loss penalizes extraneous rates while rewarding edits that move xt toward x1.
8. DNA enhancer generation (unconditional, length 500) on fly brain and melanoma ATAC-seq datasets: FlexFlow achieves the best Fréchet Biological Distance among compared diffusion/flow baselines at the same sampling budget (100 reverse steps), and ablations indicate combining a frequency-informed prior with structured coupling performs best.
9. Conditional promoter design (human promoters, length 1024) conditioned on transcription initiation profiles: FlexFlow improves MSE of predicted regulatory activity versus prior baselines, with latent guidance outperforming rate guidance (reported 0.022 vs 0.024 MSE at 100 steps), suggesting benefits from global latent steering.
10. A new peptide–MHC II conditional generation benchmark is introduced using eluted ligand data with a strict split where no 9-mer is shared across train/test clusters. On this task, FlexFlow greatly improves a held-out DeepMHCII-based discriminator score (rate guidance 0.58; latent guidance 0.66), while highlighting a quality–diversity tradeoff (latent guidance can improve plausibility while worsening embedding-distance coverage metrics).
📜Paper: arxiv.org/abs/2606.10543
#ComputationalBiology #GenerativeModels #FlowMatching #DiffusionModels #ProteinDesign #DNADesign #PeptideDesign #MHC #MachineLearning #Bioinformatics
3
16
1,420

Designing proteins starts with finding sequences that fold into the right shape.
TII’s quantum-inspired, physics-based tool generates and ranks viable sequences from the target shape alone—hundreds of times faster than standard methods.
#TII #ProteinDesign #QuantumTech

1
2
190

Jun 8
Adaptyv Bio is hiring! 25 open roles across biology, operations, software & lab automation.
Swiss “cloud lab for protein designers” building fast, automated experimental workflows and standardized protein data.
🔗 adaptyvbio.com/careers
#ProteinDesign #Biotech #AIBiology #LabAutomation

49

What if future medicines could be designed from scratch?
Researchers have developed custom-designed miniproteins capable of precisely interacting with GPCRs, the most widely targeted family of proteins in modern medicine.
These engineered proteins can selectively switch cellular signals ON or OFF, opening new possibilities for treating conditions such as cancer, obesity, diabetes, migraine, and pain disorders. By combining computational biology with protein engineering, this breakthrough demonstrates how next-generation therapeutics can be designed with greater precision and potentially fewer side effects.
From computer-designed proteins to future medicines, this research marks an exciting step towards programmable biology and precision healthcare.
Read the full research through the QR code in the final slide.
#ResearchHighlight #Nature #ProteinDesign #DrugDiscovery #PrecisionMedicine #SyntheticBiology #GPCR
@DrJitendraSingh
@rajesh_gokhale
@NABI_India @bric_ils @HydNiab @NIPGRsocial @NImmunology @BRIC_CDFD @DBT_NCCS_Pune @ICGEB @DBT_inStem @DBT_NBRC @RGCB_Trivandrum @DBT_IBSD @THSTIFaridabad @unescorcb @BIRAC_2012 @ICGEBNewDelhi @IndiaDST @moesgoi @CSIR_IND @CSIR_NIScPR @IITKanpur
5
7
867

Jun 8
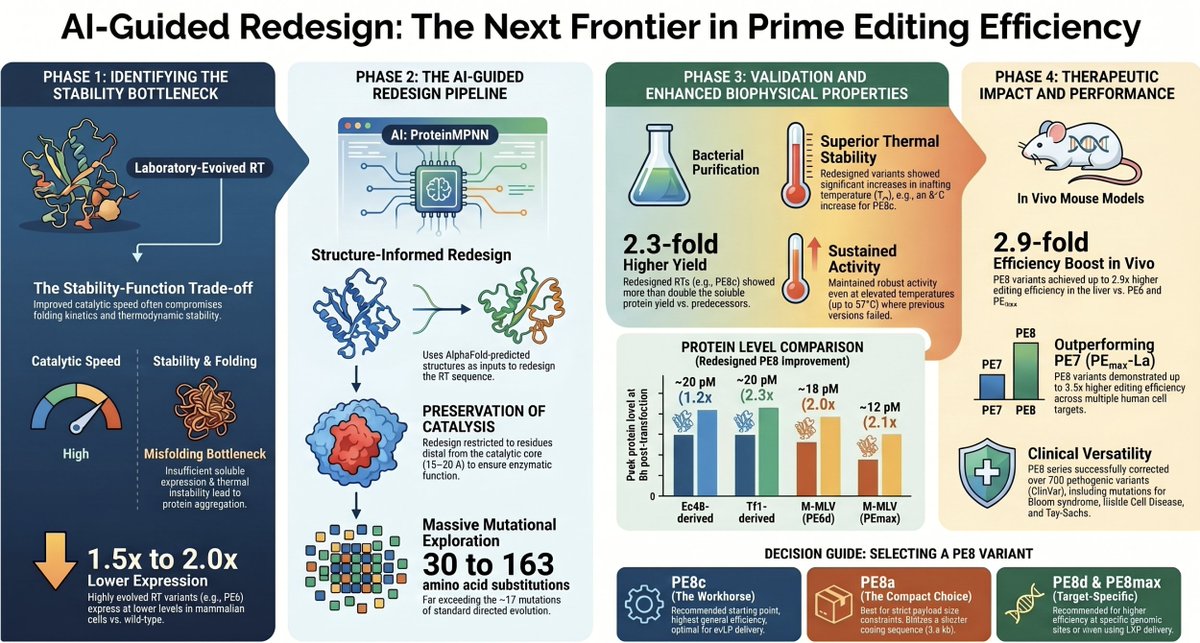
David R. Liu’s Team Upgrades Prime Editing to PE8! 🤖🧬
Published in @NatBiotech (2026): A groundbreaking fusion of AI and protein engineering overcomes the bottleneck of laboratory evolution to unleash the PE8 series! 🚀🌟
The Paradigm Shift:
1️⃣ The Evolution Trap: Lab-evolved RTs (PE6) boost catalytic activity but ruin protein folding, stability, and cellular expression levels.
2️⃣ The AI Fix: Using AlphaFold ProteinMPNN, the team reengineered the non-functional shells of RTs—boosting thermal stability by up to 8°C and doubling expression! 📈🌡️
3️⃣ In Vivo Superiority: Up to a 3.5-fold efficiency leap in primary human HSCs, T cells, and mouse livers via LNP/eVLP delivery, with zero added off-targets.
AI didn't just screen variants; it rewrote the biophysical blueprint of Prime Editors. 🏎️💨
doi.org/10.1038/s41587-026-0…
#PlantScience #GeneEditing #CRISPR #ProteinDesign #PE8


1
13
57
6,559
Heuristic multi-site optimization for protein sequence design using Masked Protein Language Models @PLOSCompBiol
1 ProtHMSO is a heuristic protein sequence design framework that uses masked protein language models (mainly ESM-2) to propose context-aware, multi-site substitutions, aiming to escape local optima and reduce the “invalid/destabilizing” variants common in blind random mutagenesis.
2 Key idea: mask one or multiple target positions, let the ProtLM output substitution probabilities conditioned on the entire sequence context, and use top-k (k=3 worked best) candidate substitutions to generate a small, high-potential mutant set for fitness scoring—shrinking combinatorial search while keeping evolutionary/biophysical plausibility.
3 The multi-site masking is central: substitutions for all masked sites are predicted synchronously from global context, so probabilities update as the sequence changes. This provides a zero-shot way to capture epistasis (synergistic residue interactions) without explicit structural supervision or task-specific training.
4 ProtHMSO is positioned as both (a) a standalone iterative optimizer and (b) a plug-in mutation operator that can replace random exploration steps inside classic search methods, improving convergence and sample-efficiency.
5 GA-HMSO: integrates ProtHMSO into a genetic algorithm by replacing random mutation with ESM-2-guided mutation, and uses a multi-objective fitness (sum of predictor scores). A dynamic schedule (higher mutation early, lower later) improved exploration–exploitation balance and avoided premature convergence.
6 MCTS-HMSO: integrates ProtHMSO into Monte Carlo Tree Search by using ESM-2 probabilities to guide expansion. It also introduces grouping of child nodes by mutation position (choose site first, then substitution), mitigating the wide-and-shallow tree problem in high-dimensional sequence action spaces.
7 AMP benchmark (DBAASP-derived; three challenging cases): across 1–5 site mutations, ProtHMSO consistently improved antimicrobial metrics (PAMP, PMIC) over random mutagenesis, while also improving structural plausibility proxies (higher ESMFold pLDDT, lower ProGen2 perplexity). Notably, random multi-site mutation degraded plausibility as sites increased, while ProtHMSO improved it.
8 ProteinGym benchmark (long proteins; single-site focus for scalability): on the Clinical substitution benchmark, ProtHMSO produced about 2x more non-pathogenic variants than random mutation at matched library sizes (10/50/100 variants per sequence). On DMS benchmarks (including targeted functional-site mutagenesis), ProtHMSO showed higher enrichment of experimentally top-ranked high-fitness mutants (top-10/20/50 overlaps) for both single- and two-site settings.
9 Practical framing: ProtHMSO acts as a high-throughput “candidate narrowing” layer—reducing millions of possibilities to tens/hundreds—while remaining compatible with adding stricter downstream filters (e.g., Rosetta/MD) in a coarse-to-fine pipeline.
💻Code: github.com/chen-bioinfo/Prot…
📜Paper: doi.org/10.1371/journal.pcbi…
#ProteinDesign #ProteinEngineering #ProteinLanguageModels #ESM2 #DirectedEvolution #GeneticAlgorithms #MCTS #AntimicrobialPeptides #ProteinGym #ComputationalBiology

1
25
2,046
AlloGen: Conformation-Selective Binder Generation with Differential State Scoring
1. AlloGen targets a core limitation in protein binder design: optimizing affinity to a single receptor structure can yield binders that engage both active/inactive (apo/holo) states, providing little functional specificity for allosteric systems (kinases, nuclear receptors, GPCRs).
2. The framework decouples generation from evaluation: any backbone generator proposes candidates for the desired state, then a learned scorer Qθ ranks or guides designs by a differential selectivity margin between goal (holo) and undesired (apo) conformations.
3. Qθ is an SE(3)-invariant interface graph transformer that scores receptor–binder interface geometry in a rigid-motion-invariant way, using interface graphs (8 Å cutoff) with residue-local frames, geometric edge features (distance RBFs, directions, relative orientations), and optional ESM-2 embeddings.
4. Training uses a two-phase curriculum to avoid degenerate “ignore-the-conformation” solutions: Phase 1 regresses to DockQ (interface quality grounding), then Phase 2 applies paired InfoNCE fine-tuning on (holo, apo, binder) triplets with cross-target negatives to force true conformational discrimination rather than receptor identity bias.
5. On 8 held-out OOD targets, Qθ shows consistent rank correlation with DockQ (mean Spearman ρ ≈ 0.520), while contact/energy proxies (PRODIGY, interface size, edge density) largely fail to track docking quality and cannot provide a differential state signal.
6. Qθ appears to encode target- and conformation-specific information: cross-target scoring shows strong diagonal dominance (designs score best on their intended target/state), and on calmodulin it produces a monotonic score increase along an interpolated apo→holo conformational path, suggesting it learns a continuous landscape rather than a binary label.
7. Because Qθ is differentiable and generator-agnostic, it supports multiple integration modes without retraining generators: passive best-of-K reranking and active guidance (classifier guidance, twisted diffusion sampling, SMC resampling, and post-generation Langevin refinement).
8. Across 15 generator×guidance combinations (RFdiffusion, PXDesign, Proteina-ComplexA), resampling-based guidance (TDS/SMC) is broadly strongest; Langevin refinement helps structure-only generators but can harm sequence-aware priors (e.g., PXDesign), emphasizing that guidance interacts with generator assumptions.
9. Experimental validation on calmodulin (a challenging ~30 Å apo↔holo rearrangement) supports the computational selectivity signal: 5/10 synthesized de novo peptides bound holo CaM (KD 46.6 nM to 1.06 µM) with no detectable apo binding, while a low-∆q negative control showed no binding—linking predicted differential scoring to measurable state specificity.
10. The study positions conformational selectivity as a learnable, transferable design objective: a modular scorer trained on paired states can retrofit existing binder-generation pipelines to design molecules that recognize functional states rather than static structures.
💻Code: huggingface.co/ChatterjeeLab…
📜Paper: arxiv.org/abs/2606.05474
#ComputationalBiology #ProteinDesign #GenerativeAI #MachineLearning #StructuralBiology #Allostery #ProteinEngineering #DiffusionModels #GNN #Calmodulin

7
35
2,384

Dr. @Green_Ahn will join @UWBiochemistry and @UWproteindesign as an Assistant Professor in January 2027. She is the first in a planned cohort of new investigators in #ProteinDesign.
👋 Welcome! bit.ly/4dP8nL1

1
8
1,039
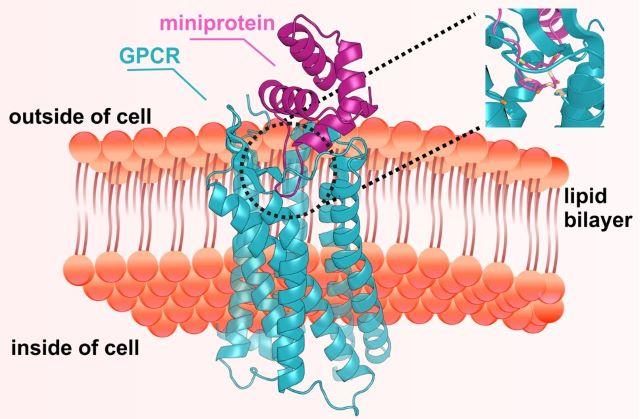
The @UWproteindesign and Skape Bio led a new study showing for the first time that AI can be used to create computationally designed proteins to activate or block GPCRs.
🧪 Abstract: go.nature.com/4ucQ3k4
📰 News: bit.ly/4x32Hon
#ProteinDesign
4
99

May 27
THE THESIS
Biohub just dropped two announcements that, read separately, look like a protein model release and a funding commitment. Read together, they're a blueprint for how AI transforms a scientific domain — and it's the opposite of what most enterprise AI strategies assume.
The real story: the bottleneck in AI-for-science isn't model intelligence. It's data. And Biohub committed $500M today to solve that bottleneck in the open.
THE EVIDENCE
ESM Cambrian (ESMC): the fourth generation of Evolutionary Scale Modeling. 300M, 600M, and 6B parameter protein language models. 300M and 600M are open-weight. The 600M model rivals ESM2's 3B. The 6B outperforms the best ESM2 by a wide margin. All trained on protein sequences using masked language modeling — the model predicts missing amino acids from context, and learns structure and function without ever being shown either.
Same principle that made GPT powerful: unsupervised learning at scale, where capabilities emerge from predicting the next token. ESMC applies it to biology. The model doesn't see protein structures during training. It learns them from patterns evolution left in sequences — because evolution can only select mutations consistent with biological function. The shadows encode the object.
The Virtual Biology Initiative: a $500M, five-year commitment to build the data foundation for predictive models of the human cell. $100M for external research coordination. $400M for data generation and next-gen measurement tech. Partners: Allen Institute, Arc Institute, Broad Institute, Wellcome Sanger, Human Cell Atlas, Human Protein Atlas. NVIDIA as compute partner.
These aren't two announcements. ESMC is what you can do with existing data. The VBI is about creating orders of magnitude more data so the next models can do vastly more. The model is the proof of concept. The initiative is the production system.
THE SO WHAT
1) Data as infrastructure, not byproduct. Every enterprise AI strategy talks about models. Biohub is building data. The Virtual Biology Initiative is the Human Genome Project for cellular measurement — coordinated, open, designed to create a shared resource no single institution could build alone.
2) Open weights as competitive strategy. Release the base openly, build the application layer commercially. Meta did it with LLaMA. Biohub is doing it for protein biology. Open weights attract researchers. Researchers produce applications. Applications create demand for frontier commercial models. It compounds.
3) Scaling laws that don't plateau. ESMC shows linear scaling from 300M to 6B with non-diminishing returns. The VBI exists because the scaling laws say "more data will work." Someone just needs to generate that data.
4) Lab-validated drug design. Researchers used ESM models to design protein binders for cancer and immune targets that effectively reactivated immune cells in lab tests. Not in silico staying in silico. Physically validated.
We've spent two years watching AI optimize the model layer. Biohub's announcement is a reminder that the leverage isn't always in the model — sometimes it's in the data infrastructure that makes the next model possible.
You need ore. Right now, for most scientific domains, we're mining with teaspoons. Biohub just committed to building the mine.
#AIDrugDiscovery #ESMC #VirtualBiology #ProteinDesign
3
63
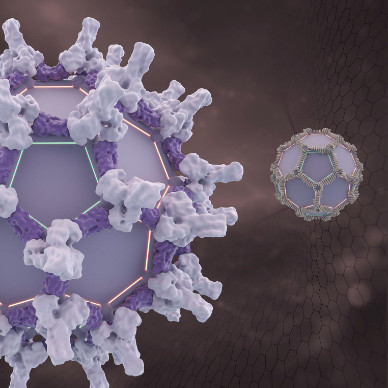
Dr. Shunzhi Wang and researchers in the Baker lab at @UWproteindesign have created a way to build virus-like protein cages that could one day serve as delivery capsules for life-saving payloads.
Read more: bit.ly/4vdlawI
#ProteinDesign

2
84
LineageFlow: Flow Matching for High-Fidelity Family-Aware Protein Sequence Generation
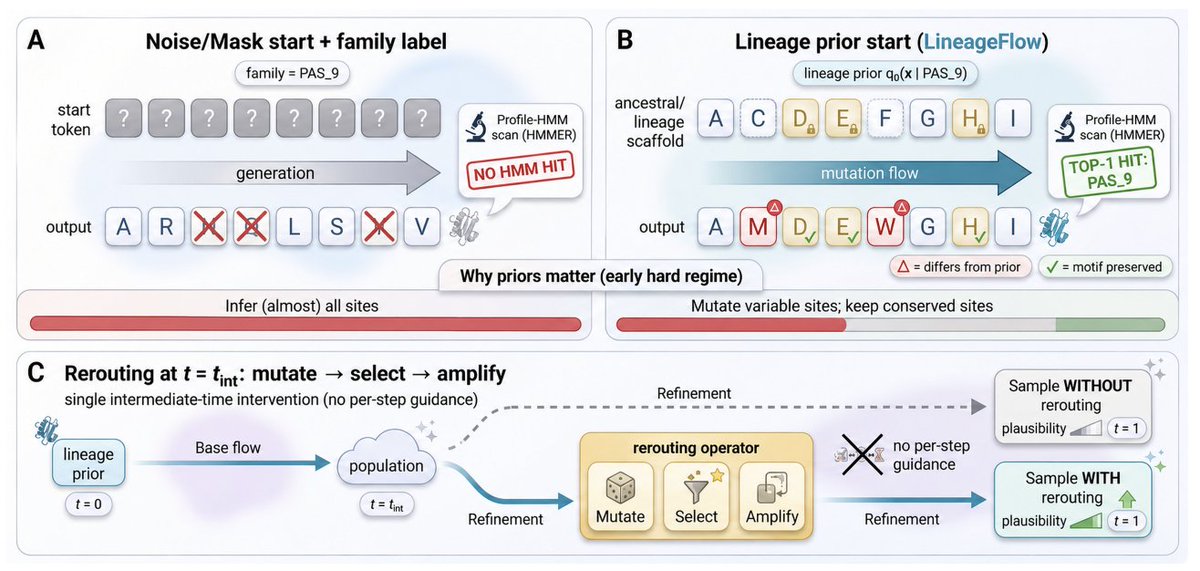
1. The paper argues that a key bottleneck in family-conditioned protein generation is the initialization prior: uniform-simplex noise or mask corruption erases evolutionary structure, forcing models to reconstruct conserved motifs “from scratch,” which weakens family control and plausibility.
2. LineageFlow replaces generic priors with lineage priors derived from ancestral sequence reconstruction (ASR): for each Pfam family, it infers a phylogeny from the MSA, performs marginal ASR at the root, and converts the site-wise root posterior into Dirichlet parameters used as a family-specific prior over the probability simplex.
3. With this design, generation is reframed as structured mutation from an evolved scaffold: conserved positions start concentrated, while variable sites retain uncertainty, aligning the trajectory with a family-specific manifold without feeding family labels or MSA prompts into the denoiser.
4. Methodologically, it builds on Dirichlet Flow Matching (DFM) on the simplex: each site follows an analytic Dirichlet path Dir(α(h,l) (tmax t) ei), with a derived lineage-specific vector field that conserves probability mass and keeps trajectories on the simplex.
5. Training uses a classifier parameterization: a transformer denoiser (initialized from ESM2) predicts terminal residues given (Xt, t), optimized by cross-entropy on valid (non-gap) MSA positions; the drift field is reconstructed by mixing analytic per-residue fields weighted by the predicted terminal distribution.
6. A second contribution is rerouting: a single intermediate-time inference intervention inspired by directed evolution (mutate → select → amplify) that steers samples toward a fitness objective without per-step gradient guidance, formalized as KL-regularized exponential tilting of the intermediate distribution.
7. Large-scale evaluation trains one shared model across 8,886 Pfam families (~8.94M sequences; 5% held-out per family) and scores generation by profile-HMM family validity (HMMER), foldability proxy (OmegaFold pLDDT), self-consistency (ESM-IF perplexity), novelty (MMseqs2 NN identity), and diversity (MMseqs2 clustering).
8. Results emphasize the role of priors: uniform-/mask-initialized baselines (DFM, EvoDiff) show essentially zero Pfam top-1 family accuracy under this strict HMM library scan, even when given explicit family labels; ASR prior alone (iid sampling) already yields high family validity, indicating ASR carries strong family signal.
9. LineageFlow with rerouting achieves near-natural family validity (Accfam 95.3% vs 96.6% for held-out natural sequences), improves foldability over prior-only and over several baselines (mean pLDDT 76.6), while keeping substantial novelty among foldable samples (Novelty@0.8 86.2%, Novelty@0.6 48.9%) and strong diversity.
10. A mechanistic analysis attributes gains to the “hard regime” at early times: Bayes-oracle denoising accuracy is higher under ASR priors than uniform priors when states are most corrupted, raising the recoverable signal ceiling and reducing early errors that propagate through the flow.
11. In a zero-shot enzyme case study, the denoiser is trained without three enzyme families, but priors are still built from their MSAs/trees; sampling without fine-tuning preserves motifs and novelty, and rerouting (using an unsupervised ESM2 plausibility objective) increases motif agreement and improves solubility/thermostability proxy distributions.
12. Limitations noted: reliance on high-quality MSAs and phylogenetic inference for priors; generation is tied to family alignment coordinates and does not model indels explicitly; evaluation relies on computational proxies (pLDDT, predictor-based properties) without experimental validation; rerouting adds compute and depends on the fitness function.
💻Code: github.com/Jinx-byebye/Linea…
📜Paper: arxiv.org/abs/2605.22252
#ComputationalBiology #ProteinDesign #GenerativeModels #FlowMatching #DiffusionModels #Phylogenetics #AncestralSequenceReconstruction #MachineLearning #Bioinformatics
7
32
2,856
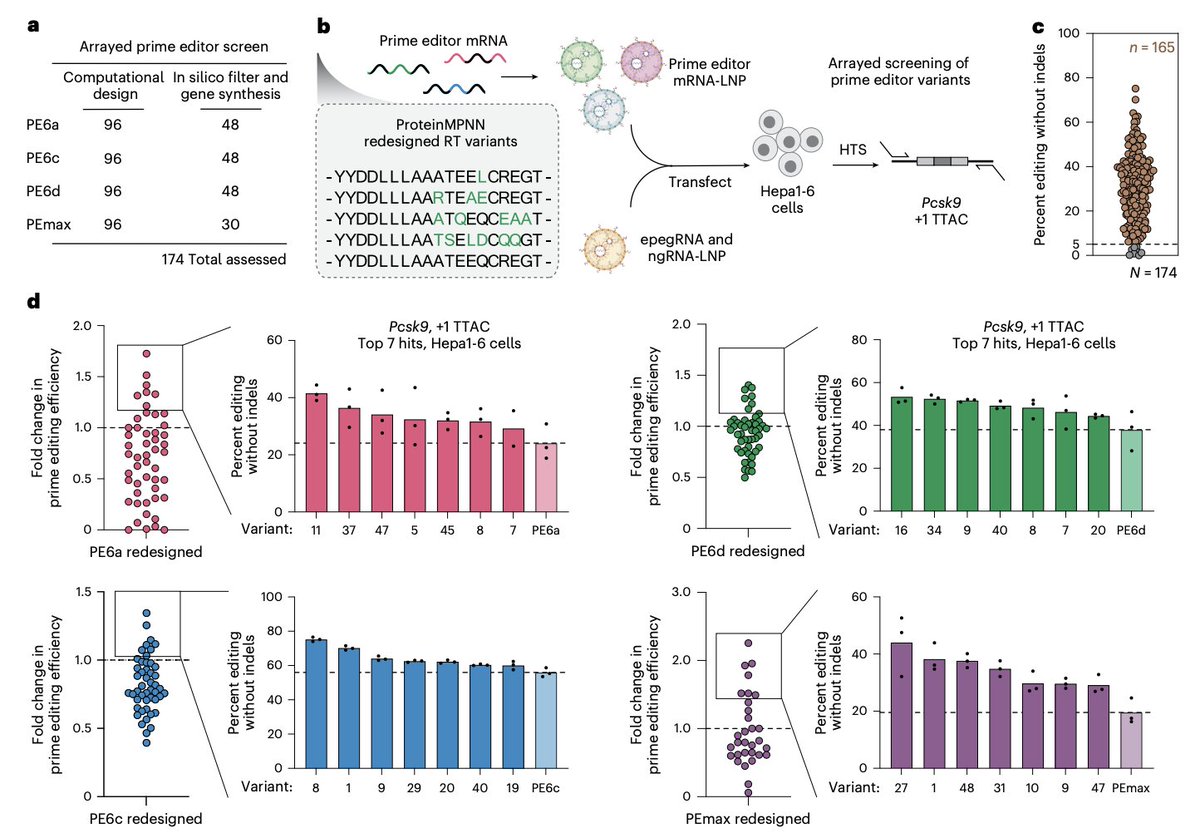
AI-guided redesign of laboratory-evolved reverse transcriptases enhances prime editing @NatureBiotech
1. The study identifies a key bottleneck in state-of-the-art prime editors: reverse transcriptases (RTs) optimized by directed evolution often trade catalytic gains for poorer folding, reduced stability, and lower intracellular expression—limiting performance especially in transient delivery settings.
2. To address this, the authors apply structure-informed AI redesign (ProteinMPNN) to RT domains from multiple prime editor lineages (PEmax, PE6a, PE6c, PE6d), while explicitly preserving residues near substrates and highly conserved positions to protect catalytic function.
3. The redesign strategy is unusually aggressive: redesigned RTs carry 30–163 amino acid substitutions (up to ~24% sequence divergence; up to ~40% of residues allowed to vary in candidate designs), far beyond prior prime editor engineering efforts that typically introduced ≤17 RT substitutions.
4. A practical pipeline is demonstrated: AlphaFold-predicted structures guide ProteinMPNN sequence generation under distance-to-substrate and conservation constraints, followed by AlphaFold2 filtering using pLDDT and RMSD to retain structurally faithful designs before synthesis and screening.
5. In an mRNA-LNP screen (Pcsk9 1 TTAC in Hepa1-6 cells), 95% of redesigned RTs remained functional (>5% editing), and ~30% outperformed their parental editors—showing that broad sequence exploration can still preserve complex RT-dependent prime editing activity.
6. Broad validation uses a pooled “self-targeting” lentiviral assay covering 700 ClinVar pathogenic variants (plus controls), totaling 16,800 prime-edit measurements across redesigned RTs; top redesigned variants improve average editing efficiency across many edits, particularly for heavily evolved RTs.
7. The best redesigned editors are named PE8 variants: PE8a (from PE6a), PE8c (from PE6c), PE8d (from PE6d), and PE8max (from PEmax). Across multiple comparisons, PE8 variants often exceed PE6/PEmax and also outperform PE7 (PEmax–La) in several tested contexts.
8. Mechanistically, redesigned RTs increase soluble expression and stability: intracellular prime editor protein levels after mRNA-LNP delivery rise up to ~2-fold (peak at ~8 h), bacterial soluble RT yields improve, and DSF shows notable thermostability gains for some designs (for example, PE8c RT 8 °C Tm over PE6c).
9. The work emphasizes that higher expression/stability translates into better editing under therapeutically relevant delivery constraints: improvements are shown in primary human fibroblasts, CD34 HSPCs, and primary T cells via mRNA electroporation, and via eVLP RNP delivery.
10. In vivo (mouse liver, Pcsk9 1 TTAC), PE8 variants increase editing up to 2.9-fold versus PE6/PEmax under modest dosing; higher in vivo protein expression accompanies higher editing, while edit:indel ratios and candidate off-target analyses remain comparable to prior editors.
💻Code: github.com/Allentaoyz/Redesi…
📜Paper: doi.org/10.1038/s41587-026-0…
#PrimeEditing #GenomeEditing #CRISPR #ProteinDesign #ProteinMPNN #AlphaFold #DirectedEvolution #mRNADelivery #LNP #ComputationalBiology
1
4
30
2,247
ConTact: Contact-First Antibody CDR Design via Explicit Interface Reasoning
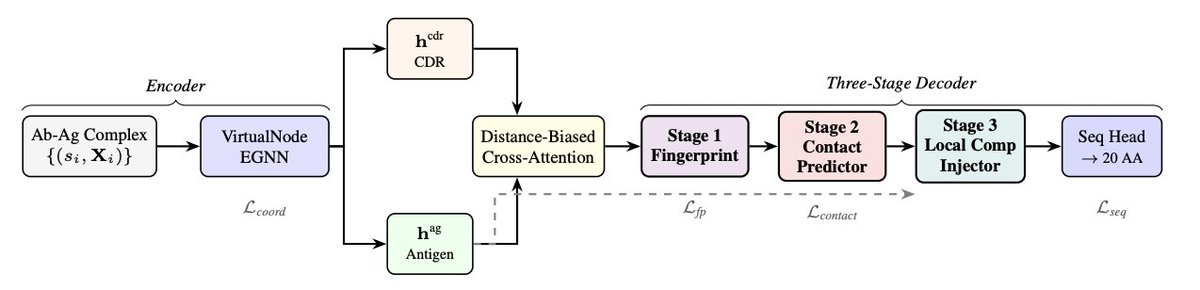
1. CONTACT reframes antigen-conditioned antibody CDR design as two distinct problems that should not be conflated: (a) deciding which CDR positions will actually contact the antigen (the “where”), and (b) choosing amino acids at those positions (the “what”). The paper argues current models often underuse antigen information because they try to solve both implicitly with uniform message passing and uniform sequence loss.
2. The core architectural idea is a contact-then-act, three-stage cascade for CDR-H3: Stage 1 learns per-position “surface complementarity fingerprints”; Stage 2 explicitly predicts which CDR residues contact the antigen (supervised); Stage 3 injects antigen features into the sequence head only where contacts are predicted, so antigen signal is routed preferentially to binding-critical positions.
3. Stage 1 (Complementarity fingerprinting) produces a compact vector per CDR position summarizing the local binding environment, inspired by molecular surface fingerprints. It is trained with an InfoNCE-style contrastive objective so positions facing similar antigen environments have similar fingerprints, improving downstream contact prediction.
4. Stage 2 (Contact prediction) uses a supervised contact label defined by a Cα–Cα threshold of 8 Å. The predictor combines CDR embeddings, KNN-aggregated antigen features, minimum-distance encodings, and the Stage 1 fingerprint. A focal binary cross-entropy loss addresses contact/non-contact imbalance and focuses learning on ambiguous boundary cases.
5. Stage 3 (Contact-guided injection) performs “double gating” to control antigen-conditioning strength at each CDR position: a learned gate multiplied by the predicted contact probability. This aims to prevent distant/noisy antigen residues from influencing non-contact positions, while still allowing fine-grained modulation at predicted contact sites.
6. The model also adds a distance-biased cross-attention module: standard cross-attention scores are augmented with a Gaussian bias based on predicted Cα distances, encoding a geometric prior that spatial neighbors should matter more for binding than far-away residues.
7. On the encoder side, CONTACT uses a heterogeneous VirtualNode-EGNN with virtual nodes connecting to all epitope residues and all CDR residues, creating a two-hop shortcut for epitope-to-CDR information flow and mitigating over-squashing that can occur when signals must traverse long chains of message passing steps.
8. Training uses a multi-term objective centered on sequence loss plus explicit contact loss and fingerprint loss, along with coordinate, pairing (CDR–antigen matching), docking (encouraging proximity to the epitope), and auxiliary regularization terms. A key detail is contact-weighted cross-entropy for sequence prediction: positions with higher predicted contact probability receive larger weights, concentrating gradient on binding-relevant residues.
9. Results on CHIMERA-BENCH (2,922 complexes; epitope-group split) show CONTACT leading on structural and interface awareness metrics among 11 retrained baselines: RMSD 1.63 Å (7% better than next-best), epitope F1 0.79 (10% over GNN baselines), fnat 0.67, DockQ 0.73, and competitive sequence recovery AAR 0.38. The paper highlights that CAAR remains low (0.20) across methods, suggesting a remaining bottleneck: Cα-level antigen representations may not capture enough chemistry (side chains/electrostatics) to nail residue identity at contacts.
📜Paper: arxiv.org/abs/2605.21600
#ComputationalBiology #AntibodyDesign #ProteinDesign #GeometricDeepLearning #GNN #EquivariantNetworks #StructuralBiology #MachineLearning
3
12
1,796