
The paper presents high-resolution X-ray structures of the Andes virus (ANDV) and Maporal virus (MAPV) Gn head (GnH) in complex with Gc, plus the homotetrameric Gn base. It highlights intra- and inter-molecular disulfide bonds that stabilize key structural elements in the prefusion Gn/Gc heterodimer and the overall square lattice on the virion surface.
Key Points on Disulfide Bonds:
• In the GnH/Gc heterodimer: The 2.2-Å structure clearly shows multiple conserved disulfide bonds (highlighted as green sticks) that stabilize the fold of both proteins and the extensive GnH–Gc interface. These help maintain the metastable prefusion conformation where Gn caps and restrains Gc’s fusion loops.
• In the Gn base (homotetramer): Disulfide bonds are shown stabilizing the protomers and inter-protomer interfaces in the membrane-proximal base, contributing to tetramer assembly and lattice organization.
• Experimental validation with "engineered" disulfides: To probe the GnH/Gc interface, researchers introduced pairs of cysteine mutations at specific contact points (marked with green bars in figures). These formed inter-chain disulfide bonds that covalently cross-linked Gn and Gc. SDS-PAGE under non-reducing conditions confirmed successful covalent linkage in virus-like particles (VLPs), validating the observed interface and demonstrating that these contacts are functionally important.
Functional Context:
These disulfides contribute to the rigid, square lattice of (Gn/Gc)₄ spikes and help control Gc fusion loop exposure/insertion during endosomal low-pH triggering. The structures also reveal evolutionary links to alphavirus glycoproteins, where similar disulfide-stabilized architectures are seen.
The discussion underscores how these bonds (both native and engineered) are critical for spike stability, lattice integrity, and preventing premature fusion—key insights for immunogen design.
#AllicinV #Allicin #DisulfideBridges #Spike #Hantavirus
pmc.ncbi.nlm.nih.gov/article…


1
3
1,005

New Preprint in collaboration with @GoogleDeepMind: AI-guided discovery of atypical protein assemblies
The @kamounlab discovered an 11-protomer complex through the Structural Novelty Index , a new way to use AlphaFold for atypical protein assemblies
buff.ly/GsF3zve
2
34
172
54,352
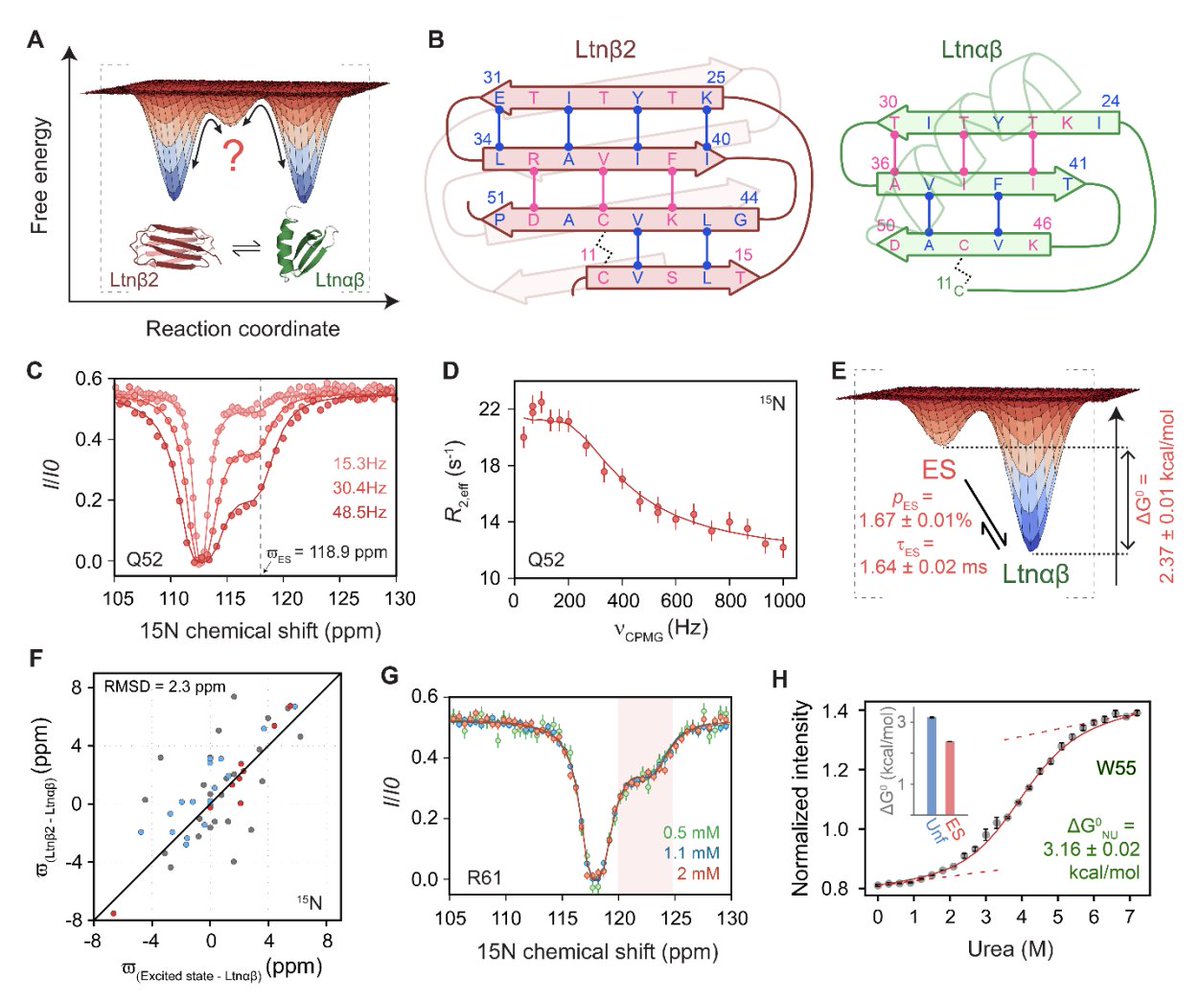
Structure of a sparsely populated chimeric intermediate that facilitates fold-switching of a metamorphic protein
1 Using multinuclear CEST relaxation-dispersion NMR, Nandi & Sekhar resolve an “invisible” excited state (ES) of the metamorphic chemokine lymphotactin (XCL1) at atomic resolution, despite it being only ~1.67% populated and ~1.6 ms-lived.
2 The key structural finding: the ES is a chimera—its secondary/tertiary structure resembles the all-β Ltnβ2 protomer, but it remains monomeric like the chemokine fold Ltnαβ. This hybrid architecture makes it a plausible bridge between two unrelated native folds.
3 The ES is not simply low-population Ltnβ2: ES vs Ltnαβ chemical-shift differences correlate poorly with the expected Ltnβ2 vs Ltnαβ shift changes, indicating a distinct conformation rather than a shifted equilibrium between the two known folds.
4 The ES is also not an oligomeric artifact: 15N CEST profiles are concentration-independent across ~0.5–2.0 mM protein, supporting a monomeric ES (important because Ltnβ2 is dimeric).
5 Multinuclear chemical shifts (15N, 13Cα, 13C’, 1HN, 1Hα) map the Ltnαβ → ES transition as a synchronized disorder/order swap: the C-terminal α-helix (54–66) unfolds (order-to-disorder), while an N-terminal segment (10–15) that is disordered in Ltnαβ becomes a β-strand (disorder-to-order), yielding a four-stranded β-sheet.
6 Chemical-shift–restrained CS-Rosetta modeling converges to an ES with four antiparallel β-strands (10–13, 25–30, 34–40, 45–51). The hydrogen-bond network matches Ltnβ2-like registry (including the characteristic single-residue shift vs Ltnαβ), but hydrophobic interface residues that would be buried in the Ltnβ2 dimer remain solvent-exposed in the ES—explaining its higher free energy.
7 Structure-guided mutagenesis validates the ES features: helix-stabilizing mutations in Ltnαβ (e.g., V59A, R61A, S62Y) reduce ES population, consistent with helix loss in ES; disrupting the putative ES N-terminal β-strand with S13P lowers ES population ~2-fold, supporting the presence of the β0 strand.
8 A decisive pathway test uses a kinetically trapped “monomorphic” chemokine-like variant (CC3; V21C/V59C) that adds a second disulfide clamp and blocks access to Ltnβ2 without strongly perturbing the Ltnαβ ground state. In CC3, the ES becomes undetectable by CEST/CPMG, supporting that the ES is on-pathway for metamorphosis rather than an off-pathway fluctuation.
9 Mechanistic implications: the authors propose Ltnβ2 must dissociate to a monomeric ES-like species, and that dimerization (burying exposed hydrophobics) is the kinetic bottleneck—fold rearrangement itself is faster. More broadly, chimeric intermediates may be a general principle enabling fold-switching and could be leveraged as a design strategy for engineered conformational switches.
📜Paper: biorxiv.org/content/10.64898…
#ComputationalBiology #StructuralBiology #NMR #ProteinDynamics #MetamorphicProteins #Chemokines #ProteinFolding #ProteinDesign #Rosetta #Biophysics
6
39
2,134

Mar 23
Yeah, Ryan's observation that mutations in 820-860 region seemed near-obligatory in "BA.3.2-like" Spikes made me wonder if this might either be about "tighter" Spike Trimer (inter-protomer binding) and/or pH-dependence of RBD opening - both features mentioned in your 2020 ref.
1
2
64

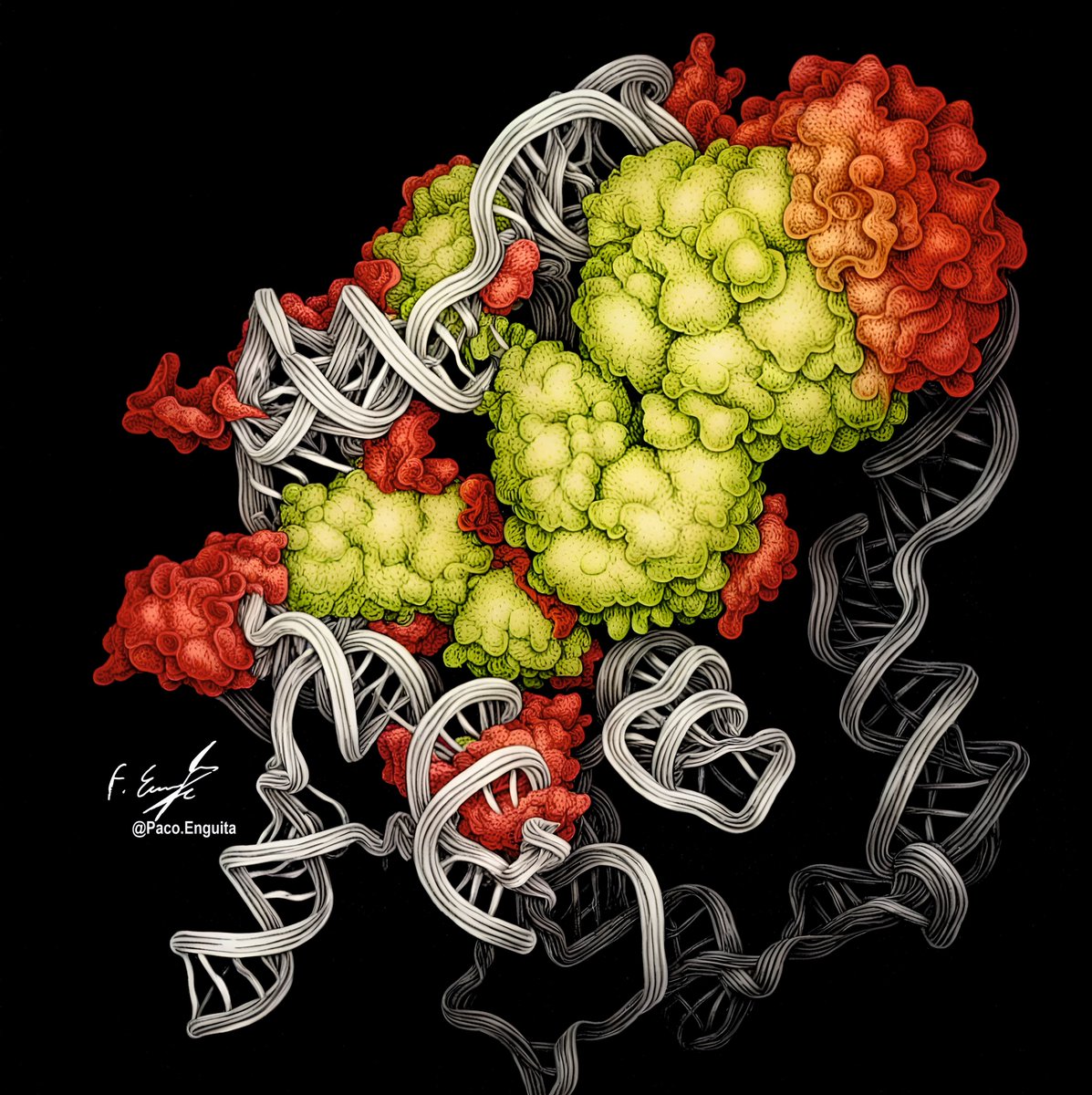
Here you can see the cryoEM structure of the H/ACA RNP protomer of human telomerase dimer (PDB code: 9QB2)
Rendering by Francisco J. Enguita (@fenguita) made with #ProteinImager
3dproteinimaging.com/protein…
#SciArt #molecularart #telomerase #complex #promoter #dimer
4
280
CryoEM structure of the Nanotube of Y5 - (5 protomer) (PDB code: 9NFN) #scivis #molecularart @dzine_ai @proteinimaging
behance.net/gallery/24496518…
5
26
633

Looking to prepare a list of SMILES for virtual screening or model training? Rowan's macroscopic pKa makes it easy to predict what the dominant protomer tautomer will be at a given pH.
Read more and see an example script in our latest blog post: rowansci.com/blog/preparing-…
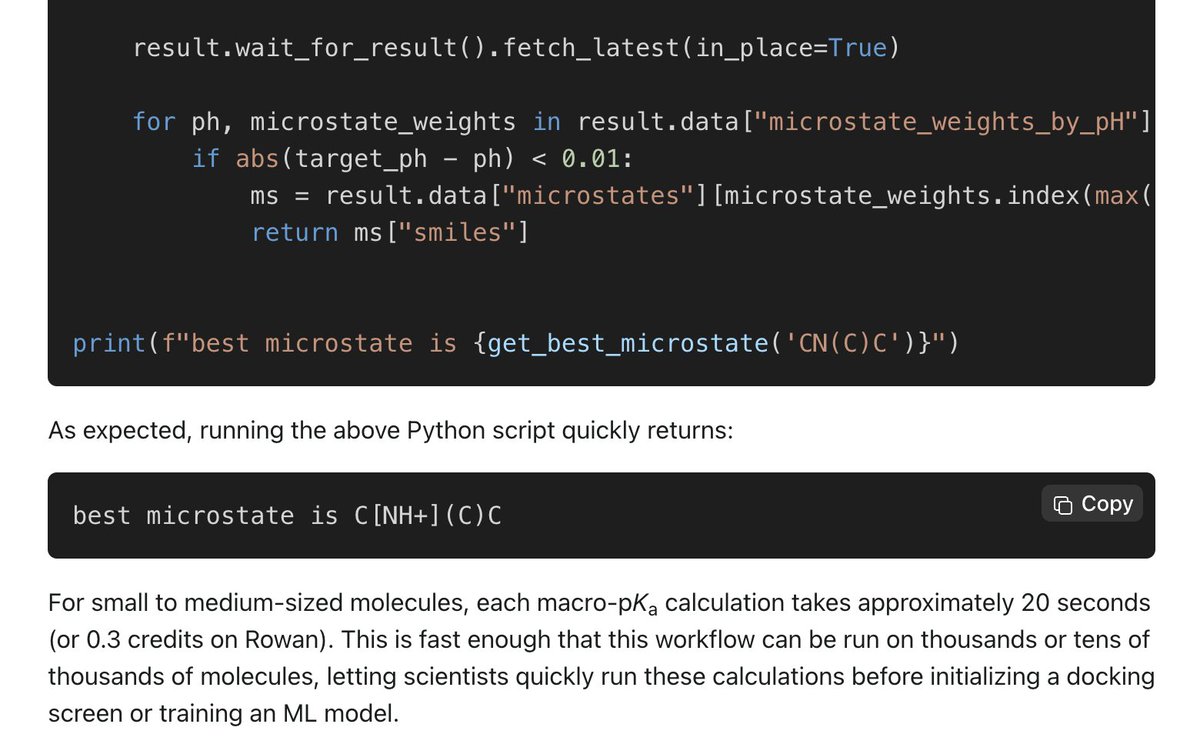
10
1,475
25 Sep 2025
Thanks!
Wondering what epitope 99N glycan is covering? There's nothing in linear sequence around there in Bloom KP.3 Ab-escape, but what else is SPACIALLY nearby?
Linked: Any chance 376 close in 3D on adjacent protomer? Might explain why 376 changed HERE, but not generally?
2
182

4 Sep 2025
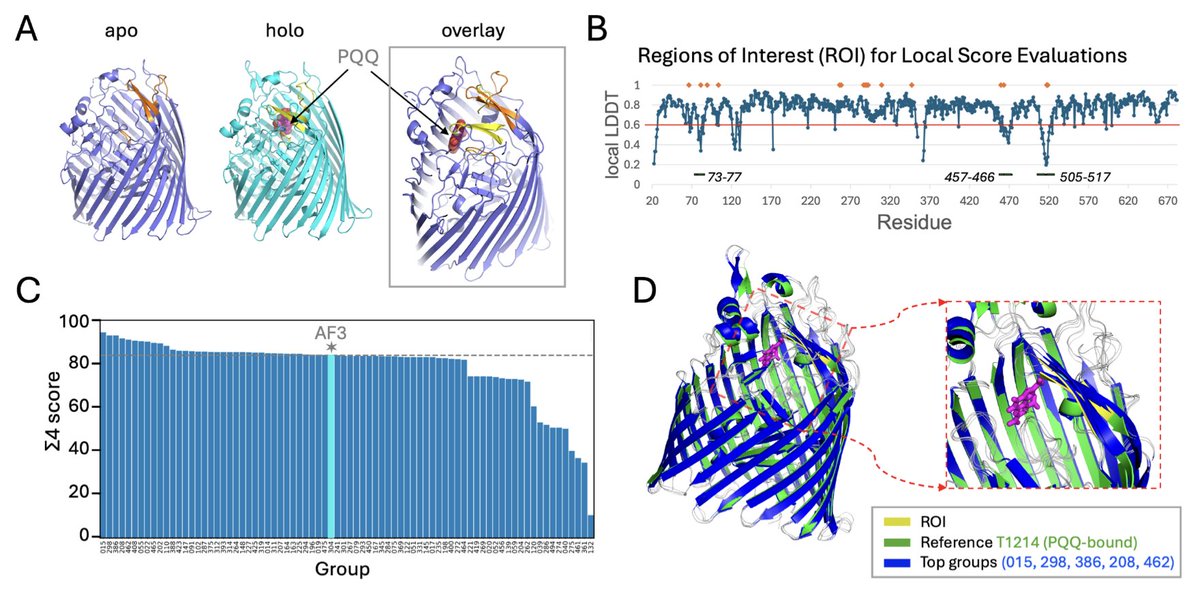
Modeling Alternative Conformational States in CASP16
1. The CASP16 Ensemble Prediction experiment assessed advances in methods for modeling proteins and nucleic acids in multiple conformational states. The study focused on targets with experimental structures determined in two or three states, evaluated by direct comparison to experimental coordinates.
2. Ten ensembles were released as community challenges, including systems with ligand-induced conformational changes, protein-DNA complexes, a trimeric protein, a stem-loop RNA, and multiple oligomeric states of a single RNA. For five targets, some groups produced reasonably accurate models of both reference states (best TM-score >0.75).
3. The most successful approaches generated multiple AlphaFold2 models using enhanced multiple sequence alignments and sampling protocols, followed by model quality-based selection. The AlphaFold3 server performed well on several targets, but individual groups outperformed it in specific cases.
4. Predictions for one protein-DNA complex, three RNA targets, and multiple oligomeric RNA states consistently fell short (TM-score <0.75). These results highlight both progress and persistent challenges in multi-state prediction. Accurate modeling of conformational ensembles, particularly RNA and large multimeric assemblies, remains a critical frontier for structural biology.
5. The study demonstrated that ligand-induced conformational changes can be captured even without explicit ligand modeling, as seen in the T1214 holo structure prediction. This suggests that MSA-driven context alone can provide sufficient information to guide the conformational shift.
6. For the M1228 target, a large serine integrase complex, predictors struggled to accurately model the tetrameric assembly and the large-scale structural change between the cis and trans states. Only a few groups captured the correct inter-dimer coiled coil (CC) handshakes and protomer conformations.
7. The T1249 target, a viral spike protein complex, presented challenges in modeling the ligand-induced interface switching between the closed and open states. While many groups achieved high TM scores, the DockQ scores indicated difficulties in accurately modeling the biologically relevant conformational change.
8. The RNA target R1203, a stem-loop structure, was particularly challenging due to the need to model non-canonical base pairs and loop regions. No group successfully predicted both states with high accuracy, highlighting limitations in current RNA prediction methods.
📜Paper: biorxiv.org/content/10.1101/…
#CASP16 #ProteinStructurePrediction #ConformationalDynamics #AlphaFold2 #AlphaFold3 #StructuralBiology #Bioinformatics
1
13
36
2,608
21 Aug 2025
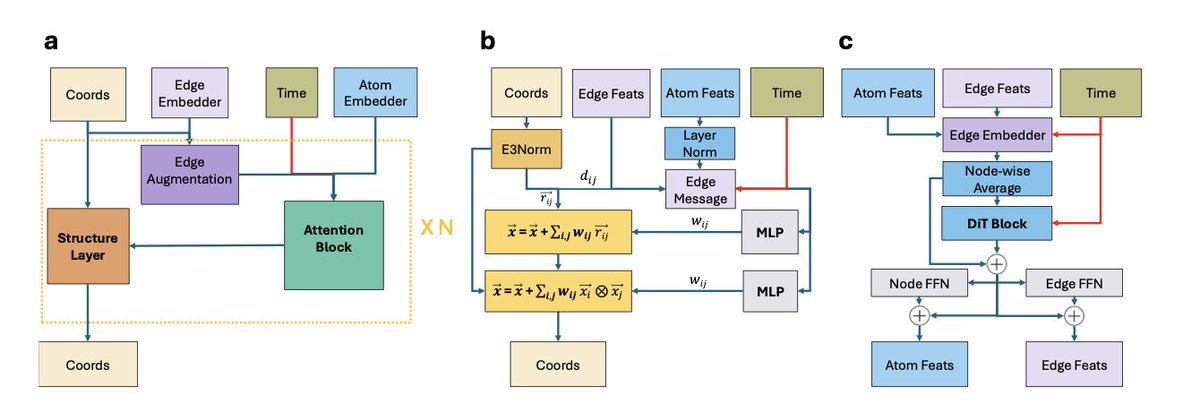
Scalable Low-Energy Molecular Conformer Generation with Quantum Mechanical Accuracy
1. Researchers have introduced LoQI, a new stereochemistry-aware diffusion model that redefines molecular conformer generation. By training on a massive, high-quality dataset, the model can generate low-energy 3D molecular geometries in a single shot, effectively integrating the conformer search and energy selection steps into a single, efficient process.
2. A key innovation is the creation of ChEMBL3D, the largest publicly available dataset of its kind. It contains over 250 million molecular conformers optimized to near-quantum mechanical accuracy. This dataset is the first to incorporate both an implicit solvent model and exhaustive stereochemistry and protomer enumeration on this scale.
3. The LoQI model significantly outperforms both traditional and other generative methods. It achieves a median relative energy of 0.33 kcal/mol, which is substantially lower than competing methods. It can also find conformers within 0.1 kcal/mol of the minimum-energy structure for a high percentage of molecules, nearly doubling the success rate of other approaches.
4. LoQI successfully addresses a significant limitation of previous generative models by explicitly encoding stereochemical information through a clever graph augmentation strategy. It achieves an accuracy for R/S and E/Z stereochemistry that is on par with traditional cheminformatics tools, making it highly practical for a wide range of applications.
5. The model proves to be highly effective at handling complex and flexible molecules, including those with a large number of rotatable bonds and macrocycles. It generates conformations that are remarkably close to true energy minima, requiring very little post-generation optimization.
💻Code: github.com/isayevlab/LoQI
📜Paper: doi.org/10.26434/chemrxiv-20…
#computationalchemistry #drugdiscovery #molecularmodeling #AIinScience #cheminformatics
4
5
1,114

18 Jul 2025
CryoEM structure of the H/ACA RNP protomer of human telomerase dimer (PDB code: 9QB2) #scivis #molecularart @dzine_ai @proteinimaging
behance.net/gallery/23062713…
5
25
609

13 Jul 2025
4/
We noticed that known anti-defense sponges of signaling molecules, despite having no sequence similarity, share key structural traits:
🔹 Small size
🔹 Homo-oligomeric assembly
🔹 Positively charged pockets at protomer interfaces
We used these features to guide our search.
1
1
5
527

23 Jun 2025
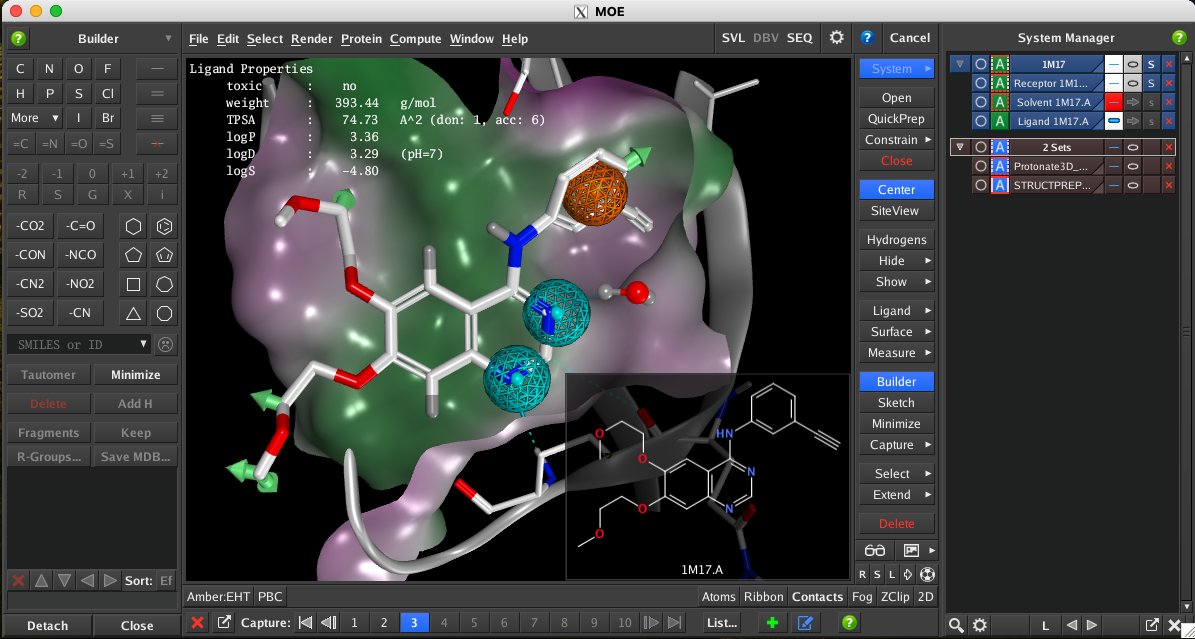
MOLECULAR OPERATING ENVIRONMENT (MOE)
Integrated Computer-Aided Molecular Design Platform
chemcomp.com/en/Products.htm
🔻3D Molecular Visualisation
🔸Easy-to-Use Graphical Interface
🔸Active Site Detection & Analysis
🔸Molecular Surfaces & Electron Density
🔸Visualize Non-bonded Interactions 🔸Publication-Quality Images & Movies
🔸GPU Accelerated 3D Stereo
Graphics
🔸Mixed Virtual Reality & 3D Printing
video.chemcomp.com/categorie…
🔻Structured-Based Design
🔸Streamlined Interface for Ligand Design
🔸Active Site Detection & Analysis 🔸Interactive Ligand Design in the Pocket
🔸Protein-Ligand Interaction Diagrams
🔸Predict Water Sites & Energetics
🔸Induced-Fit Docking Link
🔸Grow and Replace Fragments
video.chemcomp.com/categorie…
🔻Antibody and Biologics Design
🔸Structure-Based Protein Engineering
🔸Assess Liabilities & Developability 🔸Optimize Affinity, Stability & Solubility
🔸High-Throughput Antibody Modeling 🔸Generate Virtual Libraries Protein 🔸Docking and Epitope Mapping
🔸Model ADCs and Fusion Proteins
video.chemcomp.com/categorie…
🔻MOEsaic - SAR Explorer
🔸SAR and SPR Visualization
🔸Free-Wilson Compound Suggestions 🔸Matched Molecular Pairs
🔸R-Group Analysis and Profiling 🔸Substructure and Similarity Search 🔸Design Novel Virtual Compounds 🔸Document Ideas and Share Sessions
video.chemcomp.com/categorie…
🔻Ligand-Based Design
🔸Conformation Generation and Clustering
🔸Align and Superpose Small Molecules
🔸MOEsaic for SAR Exploration
🔸Pharmacophore Elucidation and Screening
🔸Generate QSAR Models – MOE Descriptors
🔸Torsion Profiles for Conformation Analysis
🔸Combinatorial Library Enumeration
video.chemcomp.com/categorie…
🔻Protein, DNA/RNA Modeling
🔸Visualize Proteins, Patches, and Interfaces
🔸Predict 3D Protein Structure from Sequence
🔸Build DNA/RNA Models
🔸Explore Mutations and Rotamers
🔸Molecular Dynamics Simulations
🔸Loop/Linker Searching and Sampling
🔸Protein-Protein Docking
video.chemcomp.com/categorie…
🔻Virtual Screening
🔸3D Pharmacophore Screening
🔸Shape and Feature Constraints
🔸Small Molecule Docking
🔸2D and 3D Fingerprint Screening
🔸Scaffold and Fragment Replacement
🔸Conformation Databases
🔸Reaction-Based Library Design
video.chemcomp.com/categorie…
🔻Fragment-Based Discovery
🔸Scaffold Hopping
🔸Fragment Linking and Growing
🔸Medicinal Chemistry Transformations
🔸Combinatorial Library Enumeration
🔸Multi-Fragment Search
🔸Ligand Hybridization (BREED)
🔸Custom Fragment Libraries
video.chemcomp.com/categorie…
🔻Structural Bioinformatics
🔸Multiple Sequence and Structure Alignment
🔸Annotate 3D Properties onto Sequences
🔸Create and Search Protein Family Databases
🔸Mine Structural Data
🔸Analyze Conserved Residues
🔸Generate Clustered Phylogenetic Trees
🔸Antibody and TCR Structural Databases
video.chemcomp.com/categorie…
🔻Molecular Simulations
🔸Molecular Mechanics and Dynamics
🔸Automated Structure Preparation
🔸Free Energy Calculations
🔸Flexible Alignment of Multiple Molecules
🔸Conformational Analysis – LowModeMD
🔸Torsion Scan and Analysis
🔸QM-Based NMR, IR and VCD Spectra
video.chemcomp.com/categorie…
🔻Peptide Modeling
🔸Macrocyclic and Linear Peptides
🔸Identify Peptide-Protein Contacts
🔸Conformational Searching
🔸Enumerate Non–Natural Peptide Libraries
🔸Structure-Based Peptide Design
🔸Optimize Peptide Properties
🔸Peptide Docking
video.chemcomp.com/categorie…
🔻Structural Biology
🔸Plot Electron Densities and Difference Maps
🔸Display Crystal Lattices and Contacts
🔸Prediction of Water Positions
🔸Electron Density-Guided Docking
🔸Create Aligned Protein Family Databases
🔸Homology Modeling for Molecular Replacement
🔸Health-Check of Protein Structures
video.chemcomp.com/categorie…
🔻Cheminformatics and QSAR
🔸400 2D and 3D Molecular Descriptors
🔸pKa Prediction and Protomer Generation
🔸Linear QSAR/QSPR
🔸Bayesian Classification / Machine Learning
🔸MOEsaic – Matched Molecular Pairs
🔸Focused Combinatorial Library Design
🔸Chemical Similarity, Diversity & Clustering
video.chemcomp.com/categorie…
1
11
15
578

7 Mar 2025
Its ridiculous to think that it wasn't announced to any of the group's official SNS and surprise the fans with that sudden announcement from the protomer than the actual company or official SNS (?) six years and seems like the company never learns and the fans were blowing up now
2
74


22 Nov 2024
Read full 🧵for explanation, but the short story is that the best apparent escape mutations all interact w/something else—like a nearby spike protomer or other important AA—making mutations there prohibitively costly.
In short, the virus has mutated itself into a corner. 2/6
2
13
114
6,903

21 Nov 2024
Answer requires thinking about full spike, not just RBD. Sites 403, 405, 504 & 505 near inter-protomer interface in down RBD structure. Most mutations there actually *increase* neutralization by sera in full-spike pseudovirus deep mutational scanning (nature.com/articles/s41586-0…)
1
7
55
2,626

30 Aug 2024
Insights into Allosteric Inhibition of the AcrB Efflux Pump: Role of Distinct Binding Pockets, Protomer Preferences, and Crosstalk Disruption #Allostery #MolecularDynamics
pubs.acs.org/doi/10.1021/acs…
#JCIM Vol64 Issue15 #compchem
8
12
1,220
18 Aug 2024
Now for Omicron: we know from structure studies & cryo-EM that Omicron (& esp. BA.2 onwards) have TIGHTER packing of RBD's & more CLOSELY-bound Spike trio generally (more inter-protomer interactions). Omicron's signature RBD 371/3/5 group contribute, as may S2 set (e.g. N969K).
1
2
75

3 Aug 2024
good part of me hates how Summer event has to be intertwined with anything of interest like ever since Full Power BB it's just been getting worse with each year
like hell I'd love a full introspect on ProtoMer, Tonelico and FGO Ciel but half their writing is now bound to Summer
112