
Ground state estimation proceeds via a classical optimizer executing the Variational Quantum Eigensolver (VQE) algorithm:
$$\min_{\boldsymbol{\beta}, \boldsymbol{\gamma}} \langle \Psi(\boldsymbol{\beta}, \boldsymbol{\gamma}) | \hat{H}_{\text{eff}}(P) | \Psi(\boldsymbol{\beta}, \boldsymbol{\gamma}) \rangle$$
3. Deep-Dive Hybrid Software Pipelines, The structural implementation of this multi-tier architecture relies on an integrated, modular open-source software pipeline. The fundamental challenge of a hybrid multi-physics model is translating sub-angstrom atomic displacements (classical structural space) into many-body qubit operators (quantum Hilbert space) via adiabatic data transfer and self-consistent loop mechanics.
Step 1: Classical Neural Net Structural Relaxation, DPmoire initializes an unrelaxed atomic supercell of $N$-layer $\text{MoTe}_2$ dictated by twist $\theta$ and a sliding vector $\mathbf{s}$. External hydrostatic pressure is applied by changing the boundary conditions of the simulation cell via an auxiliary stress tensor $\sigma_{\alpha\beta} = -P\delta_{\alpha\beta}$. The neural network uses smooth deep learning descriptors to compute the total enthalpy gradients:
$$\mathbf{F}_i = -\nabla_{\mathbf{r}_i} E_{\text{MLFF}} - P \frac{\partial V}{\partial \mathbf{r}_i}$$
The coordinates are relaxed via conjugate gradient descent until $|\mathbf{F}_i| < 10^{-4} \text{ eV/Å}$.
Step 2: The Multi-Physics Continuum Link. Once relaxed, the program extracts two separate datasets from the same structural snapshot:
1 Vibrational Hessians: The atomic positions are passed to PARPHOM to construct the $3N \times 3N$parallelized dynamical matrix $D(\mathbf{k})$, capturing how pressure hardens out-of-plane breathing modes.
2 Electronic Interlayer Registries: The localized out-of-plane distances $D_z(\mathbf{r}, P)$ between the moiré interface layers are mapped spatially. Because interlayer tunneling elements scale exponentially with distance, the pressure-dependent hopping terms are updated across the moiré superlattice cell using localized projection:
$$w(\mathbf{r}, P) = w_0 \cdot \exp\left(-\frac{D_z(\mathbf{r}, P) - D_z^{(0)}}{\lambda}\right)$$
Step 3: Low-Energy Single-Particle Diagonalization & Operator Mapping
The single-particle continuum Hamiltonian $\mathcal{H}(\theta, \mathbf{r}, P)$ is assembled into a massive, non-interacting sparse matrix. The single-particle bands are diagonalized over a grid of $\mathbf{k}$-points in the moiré Brillouin zone. If the bandwidth $W$ of the highest valence band is minimized ($W \ll U$, where $U$is the Coulomb interaction energy), the single-particle Bloch functions are projected into Maximally Localized Wannier Functions (MLWFs), $\phi_i(\mathbf{r})$.
The full many-body Hamiltonian requires calculating the matrix elements of the screened Coulomb interaction $V(\mathbf{r} - \mathbf{r}')$ using these Wannier functions:
$$V_{ijkl}(P) = \int d\mathbf{r} d\mathbf{r}' \phi_i^*(\mathbf{r})\phi_j^*(\mathbf{r}') V(\mathbf{r} - \mathbf{r}') \phi_k(\mathbf{r}')\phi_l(\mathbf{r})$$
Because $P$ compresses the lattice, the Wannier orbitals become more localized, altering the density-density interaction terms $V_{10}$ and $V_{11}$. Page 4 of 12
1
1
1
21
are scaled by parameters evaluated via database-driven quadratic regressions from density functional theory (DFT):
$$D_z(P) \approx D_z^{(0)} - \alpha_D P \beta_D P^2 \quad [\text{e.g., } 6.91 - 0.33P 0.007P^2 \ \text{Å}]$$
$$w(P) \approx w^{(0)} - \alpha_w P \beta_w P^2 \quad [\text{e.g., } -8.35 - 1.58P 0.25P^2 \ \text{meV}]$$
The spatial dependence of the moiré potential is altered by the local displacement vector field:
$$\mathbf{d}(\mathbf{r}) = \theta \hat{\mathbf{z}} \times \mathbf{r} \mathbf{s}$$
Topological invariants are characterized by calculating the valley Chern numbers $C_v$ via the Fukui-Hatsugai-Suzuki discretized Berry curvature method over the moiré Brillouin zone (Fukui et al., 2005):
$$F_{12}(\mathbf{k}_l) = \ln \left[ \frac{U_1(\mathbf{k}_l) U_2(\mathbf{k}_l \hat{\boldsymbol{\mu}}_1) U_1^\dagger(\mathbf{k}_l \hat{\boldsymbol{\mu}}_2) U_2^\dagger(\mathbf{k}_l)}{|U_1(\mathbf{k}_l) U_2(\mathbf{k}_l \hat{\boldsymbol{\mu}}_1) U_1^\dagger(\mathbf{k}_l \hat{\boldsymbol{\mu}}_2) U_2^\dagger(\mathbf{k}_l)|} \right]$$
$$C_v = \frac{1}{2\pi i} \sum_{l} F_{12}(\mathbf{k}_l)$$
Layer polarization $S(\alpha, \mathbf{k})$ and out-of-plane dipole moments $p_z$ are extracted from the self-consistent eigenstate projections across layer indices $l_i$:
$$S(\alpha, \mathbf{k}) = \sum_{i} |\phi_{i, \alpha, \mathbf{k}}|^2 l_i$$
2.3 Vibrations and Phonons under Pressure. Lattice dynamics are modeled within a supercell framework. The pressure-dependent dynamical matrix $D_{\alpha\beta}^{ij}(\mathbf{k})$ is formulated by computing the spatial Fourier transform of the interatomic force constants (IFCs) extracted via numerical differentiation of the MLFF potential energy surface at the relaxed coordinates (Mandal et al., 2024):
$$D_{\alpha\beta}^{ij}(\mathbf{k}) = \frac{1}{\sqrt{m_i m_j}} \sum_{R} \Phi_{\alpha\beta}^{ij}(\mathbf{R}) e^{-i \mathbf{k} \cdot \mathbf{R}}$$
where $\Phi_{\alpha\beta}^{ij}(\mathbf{R}) = \frac{\partial^2 E_{\text{MLFF}}}{\partial u_{i,\alpha}(\mathbf{0}) \partial u_{j,\beta}(\mathbf{R})}$ represents the second-order IFCs. The phonon frequencies $\omega_\lambda(\mathbf{k})$ and polarization vectors $\mathbf{e}_\lambda(\mathbf{k})$ are solved through the generalized secular eigenvalue equation:
$$\sum_{j,\beta} D_{\alpha\beta}^{ij}(\mathbf{k}) e_{j,\beta}^{\lambda}(\mathbf{k}) = \omega_\lambda^2(\mathbf{k}) e_{i,\alpha}^{\lambda}(\mathbf{k})$$
Mode-specific responses to structural compression are quantified using the mode Grüneisen parameter $\gamma_\lambda$:
$$\gamma_\lambda = -\frac{d \ln \omega_\lambda}{d \ln V}$$
2.4 Correlated Topological States via the Hamiltonian Variational Ansatz
To simulate fractional filling states (e.g., $\nu = 1/3$) within the isolated, pressure-optimized ultra-flat moiré bands, the continuum states are projected into a localized Wannier basis. This yields a single-band extended Hubbard-like parent Hamiltonian $\hat{H}_{\text{eff}}(P)$:
$$\hat{H}_{\text{eff}}(P) = \sum_{i,j} t_{ij}(P) \hat{c}_i^\dagger \hat{c}_j V_{10}(P) \sum_{j} \hat{n}_j \hat{n}_{j 1} V_{11}(P) \sum_{j} \hat{n}_j \hat{n}_{j 2}$$
where $V_{10}(P)$ and $V_{11}(P)$ denote the pressure-modulated first- and second-nearest-neighbor electrostatic interactions.
The state is prepared on a quantum co-processor using a symmetry-preserving Hamiltonian Variational Ansatz (HVA). The unitary ansatz state $|\Psi(\boldsymbol{\beta}, \boldsymbol{\gamma})\rangle$ is parameterized over $M$ alternating layers:
$$|\Psi(\boldsymbol{\beta}, \boldsymbol{\gamma})\rangle = \prod_{m=1}^{M} \left[ \hat{U}_V(\gamma_m) \hat{U}_t(\beta_m) \right] |\Psi_0\rangle$$
$$\hat{U}_t(\beta_m) = \exp\left( -i \beta_m \sum_{\langle i,j \rangle} (\hat{c}_i^\dagger \hat{c}_j \text{H.c.}) \right), \quad \hat{U}_V(\gamma_m) = \exp\left( -i \gamma_m \sum_{j} \hat{n}_j \hat{n}_{j 1} \right)$$
The initial state $|\Psi_0\rangle$ is selected as a classical charge-density wave (CDW) state corresponding to the targeted filling factor. Page3
1
1
1
18
This paper introduces a formal end-to-end hybrid classical-AI quantum-computing architecture designed to model, diagnose, and predict the behavior of pressure-tuned moiré quantum materials. By partitioning the problem into a classical AI surrogate stage (handling structural relaxations and phonon dynamics) and a quantum variational stage (solving the low-energy interacting Hamiltonian), we overcome the exponential scaling limits of classical exact diagonalization (ED) while ensuring structural precision over thousands of atoms.
2. Mathematical Framework
-------------------------------------------------------------
| 1. Structural Relaxation (Classical AI) |
| Minimize H_enthalpy via MLFF (DPmoire) under P and s |
-------------------------------------------------------------
|
v
-------------------------------------------------------------
| 2. Multi-Physics Property Extraction |
| - Pass coordinates to PARPHOM for Phonon Dynamics [D(k)] |
| - Fit Interlayer Distances D_z(P) and Hoppings w(P) to DFT |
-------------------------------------------------------------
|
v
-------------------------------------------------------------
| 3. Continuum Electronic Modeling |
| Construct K-Valley H(θ,r,P); compute Chern Numbers & Bands |
-------------------------------------------------------------
|
v
-------------------------------------------------------------
| 4. Quantum Correlated State Engine |
| Project Interactions onto Flat Bands -> H_eff(P) |
| Execute Hardware-Efficient HVA Circuit (PennyLane/Qiskit) |
-------------------------------------------------------------
2.1 Structural Relaxation under Twist, Sliding, and Pressure
To determine the equilibrium atomic configurations of a twisted multilayer assembly under macroscopic twist angles $\theta$, rigid interlayer sliding vectors $\mathbf{s}$, and external pressure $P$, we define a physics-informed MLFF enthalpy objective function (Liu et al., 2025):
$$H(\{\mathbf{r}_i\}, \theta, \mathbf{s}, P) = E_{\text{MLFF}}(\{\mathbf{r}_i\}, \theta, \mathbf{s}) P \cdot V(\{\mathbf{r}_i\})$$
where $\{\mathbf{r}_i\}$ denotes the full set of relaxed $3\text{D}$ atomic coordinates within the moiré supercell, and $V(\{\mathbf{r}_i\})$ represents the instantaneous volume of the simulation cell. The machine-learning force field energy term $E_{\text{MLFF}}$ is evaluated using a deep neural network potential trained on ab initio molecular dynamics (AIMD) datasets generated under varying out-of-plane strains, satisfying:
$$-\nabla_i H(\{\mathbf{r}_i\}, \theta, \mathbf{s}, P) = \mathbf{0}$$
2.2 Pressure-Dependent Effective K-Valley Hamiltonian, Following structural relaxation, the low-energy electronic structure at the $\pm K$ valleys is projected onto a continuum Hamiltonian. Extending the standard Bistritzer-MacDonald framework to encompass pressure dependence (Anfa et al., 2025), the single-particle effective Hamiltonian for a twisted bilayer system is expressed as:
$$\mathcal{H}(\theta, \mathbf{r}, P) = \begin{pmatrix} -\frac{\hbar^2}{2m^*(P)}(\mathbf{k}-\boldsymbol{\kappa}_-)^2 \Delta_t(\mathbf{r},P) & \Delta_T(\mathbf{r},P) \\ \Delta_T^\dagger(\mathbf{r},P) & -\frac{\hbar^2}{2m^*(P)}(\mathbf{k}-\boldsymbol{\kappa}_ )^2 \Delta_b(\mathbf{r},P) \end{pmatrix}$$; where $\boldsymbol{\kappa}_\pm$ denote the shifted moiré Dirac points, and $m^*(P)$ represents the pressure-dependent effective mass. The interlayer tunneling matrix $\Delta_T(\mathbf{r},P)$ and intra-layer moiré potentials $\Delta_{t/b}(\mathbf{r},P)$-
1
1
1
31
Pat Byrne - Glasgow West End retweeted

Pitches at Glasgow Green, Bellahouston Park and Springburn Park are among those expected to benefit 👇

1
2
2
525

Honoured to be a part of the inauguration of India's Multi-Lane Free Flow (MLFF) barrier-less tolling system at the Mundka–Bakkarwala Toll Plaza on Urban Extension Road-II (UER-II) in Delhi.
#MLFF #UERII #NHAI #HighwayInnovation #DigitalIndia #BuildingANation



8

Jun 14
🚛 Delhi plans to convert all 154 commercial vehicle entry points into barrier-less toll corridors by year-end.
📍 The new MLFF system will replace conventional toll booths across city borders.
📡 RFID technology and ANPR cameras will enable automatic toll collection without vehicles stopping.swarajyamag.com/news-brief/d…
1
15
90
4,227

مساج في الدمام
Massage in #riyadh
🍮🤸♂️🛤️ Wa.me/966548017237
مساج في #الرياض
Massage in #jeddah
🇰🇾🥗 xxxx Russian🚖📿
mLff

4

Jun 13
No MLFF on Bengaluru-Mysuru Expressway, NHAI changes course
#BengaluruMysuruExpressway #NHAI #TollUpdate #HighwayNews #KarnatakaNews
Read more:
newsfirstprime.com/bengaluru…
47

Jun 11
🧩 Quick check — can you crack this?
With reference to India's first Multi-Lane Free Flow (MLFF) barrier-less tolling system launched at the Chorayasi Toll Plaza, which one of the following is its executing/implementing agency under the Ministry of Road Transport and Highways?
(a) Indian Highways Management Company Limited (IHMCL)
(b) National Highways Authority of India (NHAI)
(c) National Highways and Infrastructure Development Corporation Limited (NHIDCL)
(d) Indian Road Congress (IRC)
👉 Tap for the answer, explanation & more questions on this topic:
api.learnthrupractice.com/sh…
#UPSC #PIB #PrelimsPractice
22
Jun 11
🛰️ what Chorayasi is really a rehearsal for
MLFF is the bridge, not the destination. NHAI's eventual plan is satellite-based, distance-linked tolling — and the architecture is being built so that 8 crore FASTag users are not disrupted on the way there.
• 2024 Global EoI invited for GNSS-based ETC
• Hybrid roll-out planned: RFID-FASTag lanes alongside dedicated GNSS lanes
• Enforcement depends on VAHAN/Sarathi data and HSRP plate integrity
1
24
Jun 11
🧭 how the system actually reads a moving car
MLFF does not replace FASTag. It layers a second pair of eyes on top of it, so a vehicle never has to pause.
• Automatic Number Plate Recognition (ANPR) cameras read the plate via OCR
• FASTag RFID debits the toll at highway speed
• A central back-end reconciles the transaction
• If FASTag is invalid or insufficient, an Electronic Notice is issued
• Payment window: 72 hours; default attracts 2× toll as penalty
1
16
Jun 11
📜 the long road to a barrier-less booth
The idea had been inching forward for a decade, one policy step at a time.
• 2016: FASTag RFID-based electronic toll collection introduced by NHAI/IHMCL
• 2019–21: FASTag made mandatory across NH toll plazas
• 2023: NHAI Hackathon on barrier-less tolling and intelligent traffic management
• 2024: Global Expression of Interest invited for GNSS-based ETC
• 2025: NHAI signs concessionaire agreement for the Chorayasi MLFF pilot
1
32
Jun 11
🗺️ the plaza that stopped stopping traffic
The setting is the Surat–Bharuch stretch of NH-48 — the old Delhi–Mumbai artery, part of the Golden Quadrilateral. It was here that Union Minister Nitin Gadkari flagged off India's first Multi-Lane Free Flow, or MLFF, system.
• Site: Chorayasi Toll Plaza, Gujarat
• Corridor: Surat–Bharuch section, NH-48
• Launched by: MoRTH, executed through NHAI
1
52
Jun 11
India’s First Multi-Lane Free Flow (MLFF) Barrier-less Tolling System in Gujarat
A Prelims favourite in the making: India's first Multi-Lane Free Flow tolling system, the technologies it rides on, and the statutory framework behind highway user fees — all GS-III infrastructure territory.
🚆 On 1 May 2026, a toll plaza in Gujarat quietly did something no Indian highway had done before. The boom barriers did not come down. The cars did not slow. And yet, by the time roughly 41,500 vehicles had passed through Chorayasi that day, every single one had paid.
learnthrupractice.com/pib-it…
#UPSC #PIB #Infrastructure
1
128

We are at least 20 years behind in toll management.
Why do we still crawl at 5 km/h for FASTag boom barriers to lift? One error, and the whole lane stalls.
Minister Gadkari had promised 80 km/h zero-wait tolling on National Highways by the end of 2026, and NHAI just launched India's first MLFF pilot in Gujarat last month.
Why stop at National Highways? Shouldn't State Highways and Expressways also adopt one uniform nationwide tolling system?
We need ONE uniform system across India.
Open Roads: Overhead steel gantries replace bulky concrete plazas.
High-speed ANPR cameras RFID scanning at full highway speeds.
10-15 day buffer to recharge before a fine hits.
Dynamic toll rates for peak vs. low-traffic hours.
Millions of liters of fuel saved by ending stop-and-start idling.

8
16
108
9,947

Jun 10
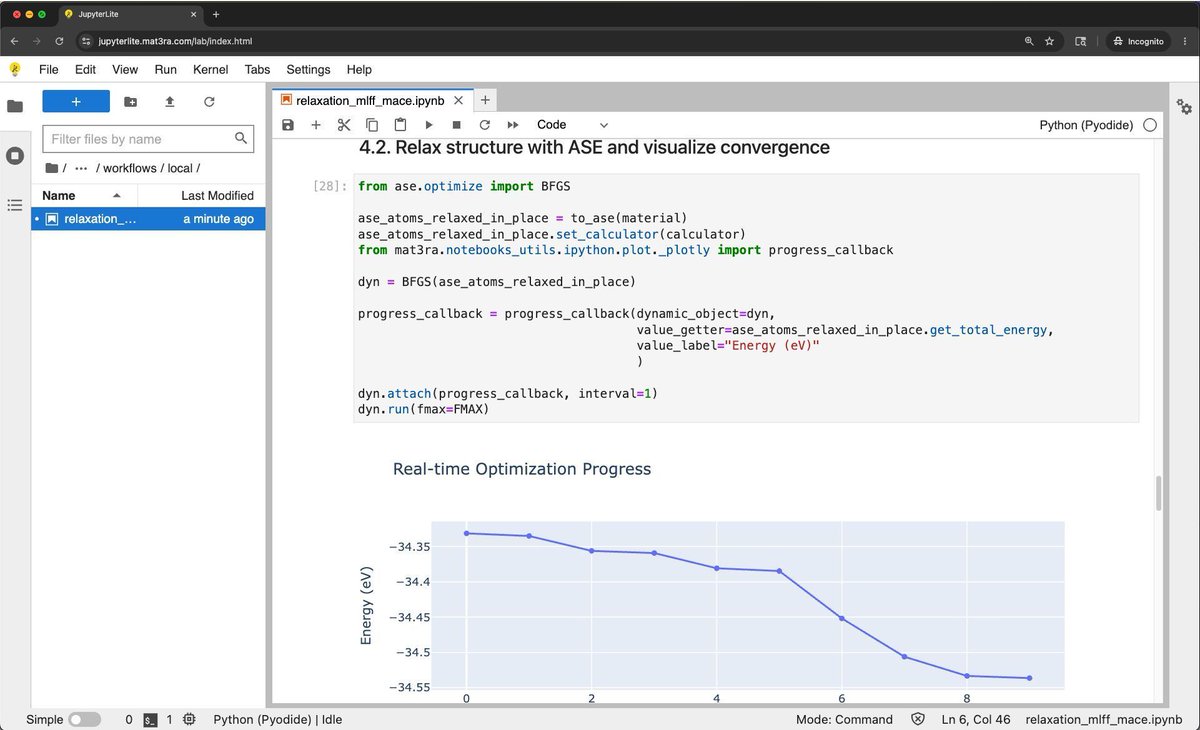
⚡️ 𝗥𝗲𝗹𝗲𝗮𝘀𝗲 𝟮𝟬𝟮𝟲.𝟱.𝟮𝟴 𝗵𝗶𝗴𝗵𝗹𝗶𝗴𝗵𝘁𝘀 - 𝗽𝗿𝗲-𝗿𝗲𝗹𝗮𝘅 𝘀𝘁𝗿𝘂𝗰𝘁𝘂𝗿𝗲𝘀 𝘄𝗶𝘁𝗵 𝗠𝗟𝗙𝗙 𝗶𝗻 𝗝𝘂𝗽𝘆𝘁𝗲𝗿𝗟𝗶𝘁𝗲/𝗣𝘆𝗼𝗱𝗶𝗱𝗲
For many researchers, the hardest part of testing a new force field is not the model itself. It is everything around it: Python environments, dependencies, runtime setup, and infrastructure.
That is why a notebook showing how to use the MACE universal forcefield in Jupyter and JupyterLite is such an interesting addition. The workflow demonstrates browser-based structural pre-relaxation with no extra infrastructure required.
This is useful because it lowers the barrier to trying MLFF-based workflows, makes the setup steps visible and easier to learn, provides a lightweight path for education, testing, and early-stage validation, and creates a practical entry point before moving to larger compute environments.
In many scientific settings, reducing the startup cost of a workflow is already a major improvement. A browser-based notebook does not replace production-scale infrastructure, but it makes the workflow easier to understand, test, and share. For anyone exploring pre-relaxation workflows before DFT, this is a very practical direction.
Release page: mat3ra.com/news-and-blog-pos…
Related webinar resource: mat3ra.com/events-posts/mat3…
👉 Try it for free at mat3ra.com/?utm_source=twitt…
#materials #RnD #mat3ra #exabyteio #materialsscience #materialsdesign #materialsmodeling #science #technology
1
106
Jun 10
💰 the quieter story underneath the launch
The Minister, Nitin Gadkari, formally launched the system at Chorayasi. But the more interesting numbers sit in the operational accounts.
• Day-1 traffic at Chorayasi: ~41,500 vehicles
• Operational cost falls from ~15% of toll revenue to ~3–4%
• Estimated system-wide savings: ₹5,000–6,000 crore a year
• Rollout plan: ~25 NH fee plazas to receive MLFF in the current financial year
• Reduced idling at plazas also trims vehicular emissions and fuel burn
1
21
Jun 10
🧭 what MLFF actually is
Multi-Lane Free Flow tolling is barrier-less and non-stop. It does not yet rely on satellites — it leans on two technologies already inside the country's vehicle ecosystem.
• Core tech: Automatic Number Plate Recognition (ANPR) FASTag (RFID)
• Cameras read the plate as the vehicle passes at highway speed
• Plate-to-owner mapping draws on the VAHAN database
• MLFF is a precursor to fully GNSS-based satellite tolling, not a substitute
1
13
Jun 10
📜 the long road from cash booth to camera
Tolling in India has shed a skin every few years, each step trying to shorten the queue.
• 2016: FASTag introduced on NH plazas — RFID-based electronic toll collection
• 1 January 2021: FASTag made mandatory for all vehicles on national highways
• 2025: NHAI signs the agreement to implement India's first MLFF system in Gujarat
• 1 May 2025: MoRTH clarifies GNSS/satellite tolling will not launch — MLFF is chosen instead
1
29