Protein-protein interaction prediction using bidirectional GRUs with explicit ensemble
@PLOSONE
1.This paper introduces a novel model for predicting protein-protein interactions (PPIs) using a combination of bidirectional gated recurrent units (BiGRUs) and an explicit ensemble approach. The model achieves state-of-the-art performance on both in-species and cross-species datasets.
2.The most attractive feature is its high generalizability. Trained on S. cerevisiae, the model maintains strong performance across multiple cross-species and disease-specific datasets—highlighting its robustness and potential utility in diverse biological settings.
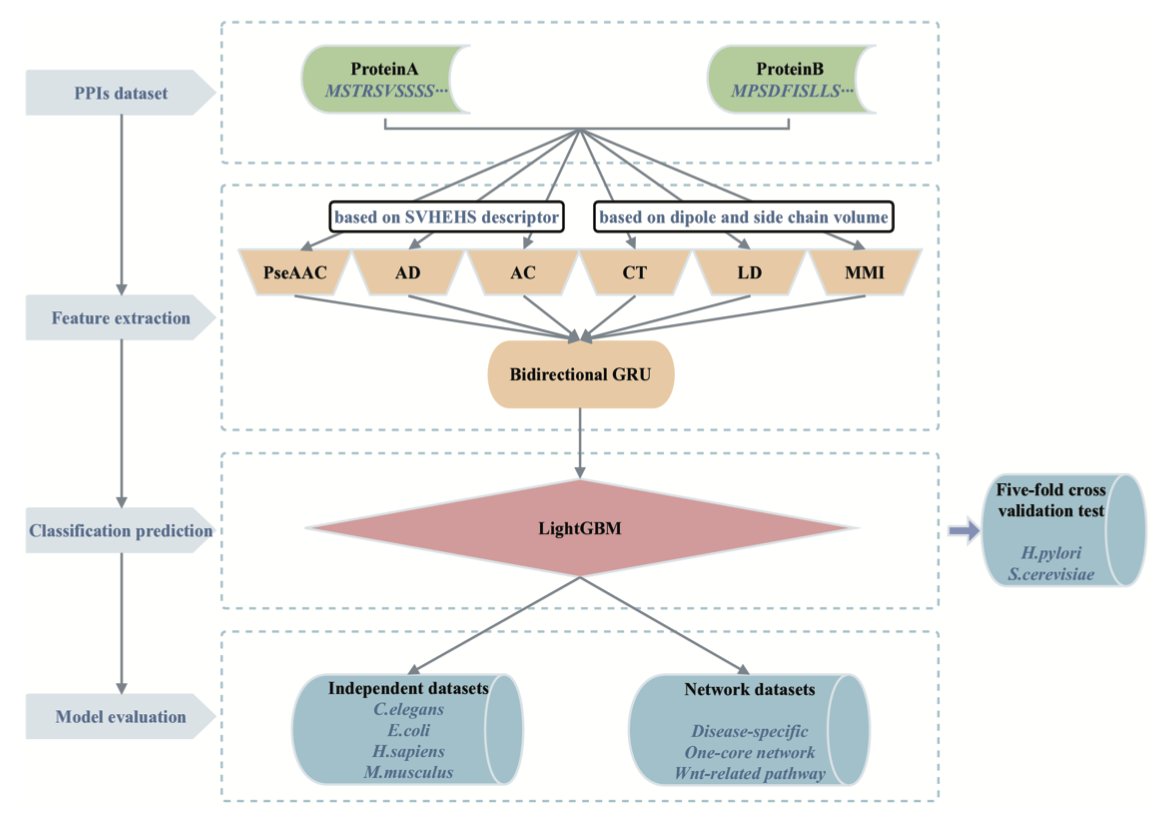
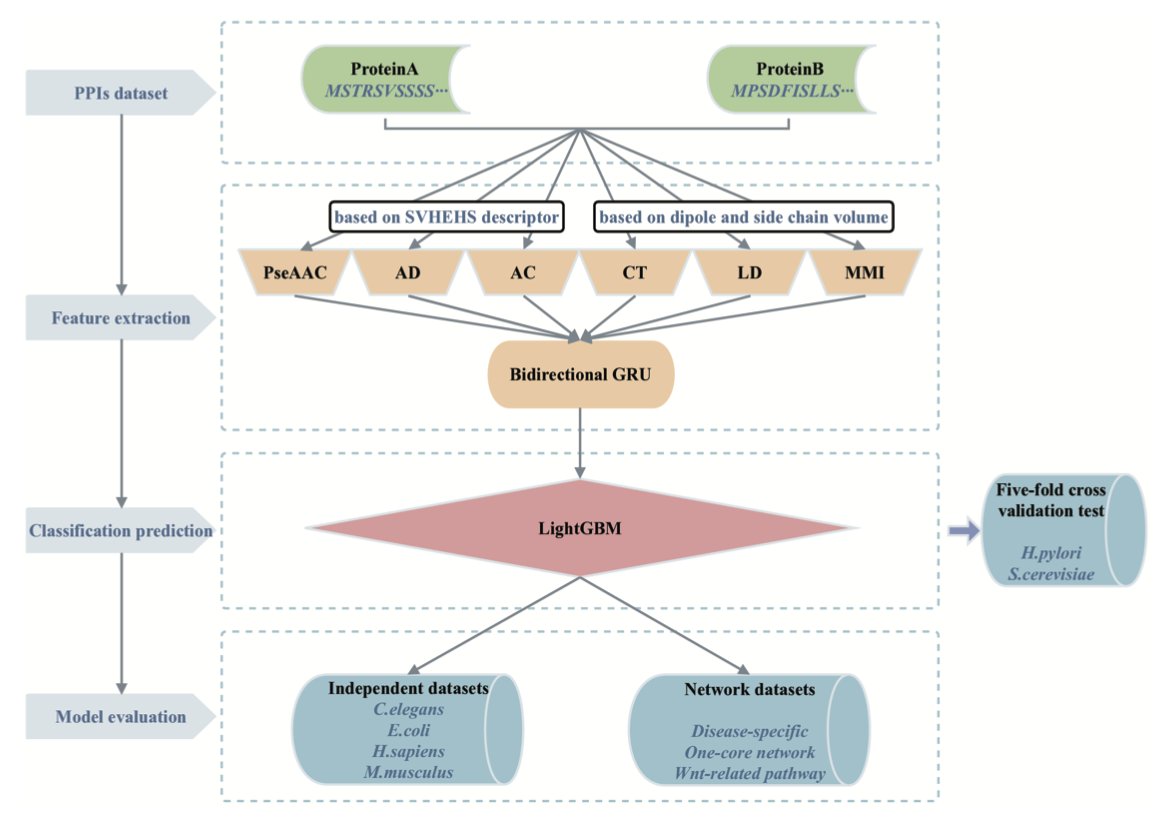
3.To enhance sequence representation, the authors incorporate the SVHEHS descriptor, a 20×13-dimensional matrix derived from 457 physicochemical properties of amino acids, into three key feature encoding techniques: PseAAC, AD, and AC.
4.Six feature coding techniques are used in total—PseAAC, AD, AC, CT, LD, and MMI—each capturing different aspects of protein sequences (composition, sequence order, local/global dependencies, and mutual information).
5.Each feature vector is processed by a dedicated BiGRU for dimensionality reduction. These BiGRUs are then explicitly ensembled to retain diverse learned features while reducing noise and redundancy.
6.The final feature set is input into a LightGBM classifier. This combination of deep learning for feature transformation and gradient boosting for classification allows for both high accuracy and efficient inference.
7.On the H. pylori and S. cerevisiae datasets, the model achieves 96.47% and 97.79% accuracy, respectively—outperforming models like GcForest-PPI, GTB-PPI, and DeepPPI.
8.BiGRU outperforms both forward and backward GRUs in capturing sequence dependencies. Bidirectional context is shown to be crucial for capturing interaction-related patterns.
9.The explicit ensemble (MultiEns) of six independent BiGRUs outperforms simpler strategies like feature concatenation (MultiCon) or dual-branch networks (MultiSep), demonstrating the benefit of architectural diversity.
10.The model also surpasses traditional classifiers (e.g., SVM, KNN, RF, AdaBoost) in accuracy, robustness, and computational efficiency, justifying the choice of LightGBM for final prediction.
11.Evaluation on independent datasets (e.g., C. elegans, E. coli, H. sapiens, M. musculus) confirms excellent generalization, with accuracy consistently above 94%. On disease-specific datasets, it achieves perfect accuracy in some cases.
12.The framework is modular and data-efficient. A key limitation is the absence of protein structural data or pretrained language model embeddings, which the authors note as future work.
13.Overall, this study presents a flexible and accurate PPI prediction pipeline with demonstrated cross-domain utility and a well-grounded methodological design.
💻Code:
github.com/bingo111111/BiGRU…
📜Paper:
journals.plos.org/plosone/ar…
#PPI #DeepLearning #Bioinformatics #GRU #ProteinInteractions #MachineLearning #BiGRU #LightGBM #SequenceAnalysis