
Viral Proteins Reveal Geometry of Protein Language Models
1. The paper shows that protein language model (pLM) embedding spaces are dominated by a single “nativeness axis” (PC1) that strongly aligns with masked-reconstruction perplexity (a model-relative measure of how in-distribution a sequence is). This axis orders sequences from well-modeled cellular proteins to viral proteins to shuffled/random controls.
2. In ESMC-600M, PC1 explains 73.1% of embedding variance and correlates with perplexity at Spearman ρ=0.961, indicating that reconstruction difficulty is not just a score but a major geometric direction organizing embeddings across the tree of life.
3. Viral proteins are not treated as extreme outliers: they sit in an intermediate region—less “native” than cellular proteins but more structured than biologically meaningless sequences (position-shuffled or i.i.d. random), suggesting pLMs encode a continuum from in-distribution to out-of-distribution.
4. The same nativeness-axis geometry generalizes across ESM families (ESM2, ESMC, ESM3), including ESM3-OPEN (trained without viral sequences), and also appears in non-ESM architectures (ProGen2 autoregressive; EvoDiff discrete diffusion). This supports the idea that a dominant “model-fit” direction may be a broader property of sequence models trained on imbalanced biological data.
5. The work quantifies the underlying imbalance: UniRef50 pretraining coverage is heavily cellular-dominated (about 46.3M cellular clusters vs 390.3k viral; ~119× ratio), motivating the question of how underrepresented viral sequences are represented.
6. A key control argues nativeness is not simply “seen vs unseen”: cellular Swiss-Prot proteins released after an ESMC checkpoint (thus absent from its pretraining data) still look far more native-like (median PPL 5.3) than human viral proteins (median PPL 15.3), implying nativeness reflects compatibility with a cellular-dominated prior more than mere exposure.
7. Scaling changes viral nativeness, but unevenly across viral families: in ESMC (300M→6B), the fraction of human viral proteins below a native-like threshold (PPL<5) increases only modestly overall (~5%→~17%), while some families (e.g., Papillomaviridae, Retroviridae) shift strongly toward the native region and others (e.g., Orthomyxoviridae, Orthoherpesviridae, Sedoreoviridae) remain displaced.
8. Despite this dominant nativeness axis, embeddings retain viral-specific information beyond perplexity: linear probes on mean-pooled embeddings classify human viral vs cellular proteins with near-ceiling AUC (0.97–1.00 for larger models) under a homology-controlled split, and outperform both perplexity-only zero-shot classification and shallow sequence baselines (length, amino-acid composition, dipeptide composition).
9. The separation is especially relevant at low false-positive rates (screening-like settings): at 1% FPR, embedding probes achieve much higher TPR than perplexity-only classifiers (e.g., ESMC-6B 96.7% vs 39.2%), showing that “viral identity” is linearly accessible even when perplexity becomes a weaker separator at large scale.
10. Implications: nativeness (perplexity / PC1 position) can act as a diagnostic for where pLMs may be less reliable (notably for certain viral families), while embedding-based signals may complement homology methods for viral detection and biosecurity screening—though the authors emphasize evaluation and safety framing over deployment.
💻Code: github.com/MisteFr/viral-pro…
📜Paper: arxiv.org/abs/2606.12609
#ProteinLanguageModels #ESM #ViralProteins #RepresentationLearning #ComputationalBiology #Bioinformatics #MachineLearning #Biosecurity #Interpretability #ScalingLaws

2
41
3,123
Constraint-Aware Optimization for Robust Protein Stability Prediction
1. The paper proposes an optimization-level framework (no architecture changes) to improve robustness of multimodal protein stability (ΔΔG) predictors built on the SPURS-style backbone (ESM2 sequence ProteinMPNN structure, fused per-residue features, MLP head with ΔΔG = score(mut) − score(wt)).
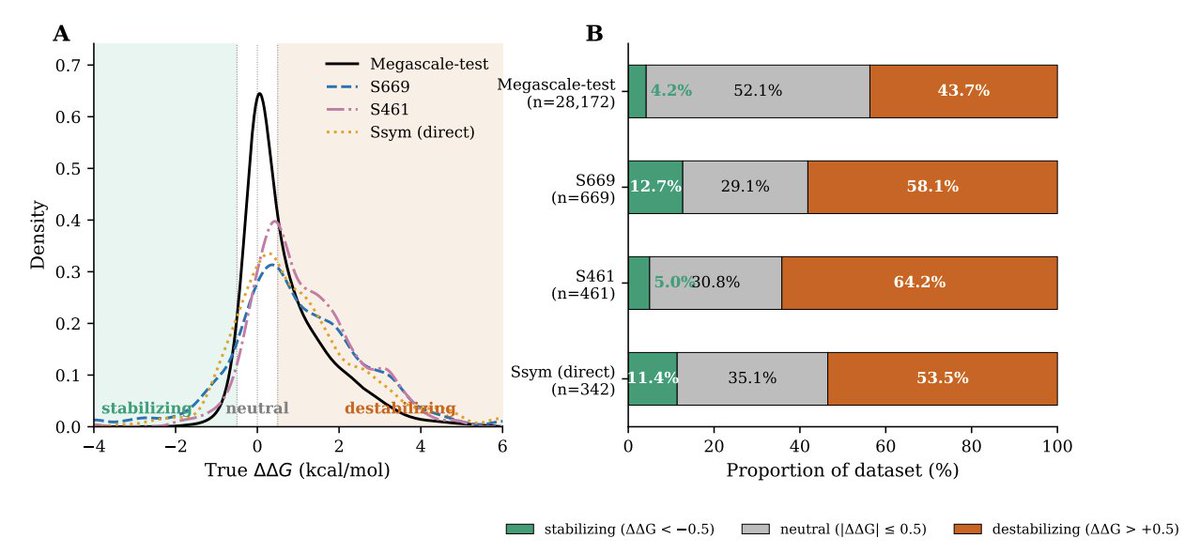
2. Core motivation: strong in-distribution performance on Megascale does not translate to out-of-distribution (OOD) proteins; datasets are heavily label-imbalanced (stabilizing mutations are rare, ~4–13% across common benchmarks), and predictors show persistent forward–reverse bias on paired-mutation tests (Ssym).
3. The framework combines three losses that target different failure modes: (i) Balanced MSE (BMC) to counter ΔΔG label imbalance, (ii) a Siamese anti-symmetric regularizer to encourage thermodynamic reversibility, and (iii) a new OOD-margin consistency loss that penalizes prediction sensitivity to small perturbations of the per-position fused representation.
4. Headline OOD results across 3 seeds and 11 benchmarks: Spearman on S669 improves from 0.486 to 0.540 (σ=0.002), and on S461 from 0.653 to 0.711. Additional smaller gains are reported on S8754, S2648, S4346, and Ssym-direct; performance drops modestly on in-distribution Megascale-test (0.749 → 0.713), interpreted as a robustness tradeoff.
5. BMC is used as a distribution-aware regression objective with a learnable noise scale, designed to increase gradient pressure on underrepresented ΔΔG regions (especially stabilizing tail) rather than letting MSE/Huber be dominated by neutral/destabilizing examples.
6. The Siamese anti-symmetric loss is applied by evaluating both wt→mut and mut→wt with shared weights and penalizing (f→ f←)^2. Ablations suggest it contributes additively with BMC on the hardest OOD sets, but it can hurt ΔTm benchmarks (e.g., S571), consistent with ΔTm not obeying the same magnitude constraints as ΔΔG.
7. The OOD-margin loss is a representation-stability regularizer: add small Gaussian noise to the fused residue representation after the encoder forward pass, re-run only the MLP head, and penalize (ŷclean − ŷnoisy)^2. It adds ~10% per-step training cost and shows a localized optimum around noise scale σ≈0.20 (too large degrades both OOD gains and in-distribution fit).
8. Mechanistic diagnostic on Ssym: anti-symmetric training does not eliminate systematic forward–reverse bias (offsets remain ~0.3–0.4 kcal/mol). The paper argues gains mainly come from implicit regularization/optimization dynamics rather than strict enforcement of thermodynamic constraints; even an explicit bias-corrected anti-symmetry loss reduces bias but does not improve OOD Spearman.
9. Practical engineering angle: for retrieving rare stabilizing mutations (ΔΔG ≤ −0.5) on S669, the combined objective improves top-50% stabilizing recall (0.659 → 0.685), suggesting better candidate yield in typical screening-style prioritization where the stabilizing tail matters more than average error near neutrality.
10. Negative results help delineate what does not help OOD here: auxiliary multitask supervision with K50 adds little (ΔΔG already highly correlated with K50), and ProteinMPNN-based structural relaxation/perturbation features did not improve key wild-type-based OOD sets (S669/S461), reinforcing that optimization behavior itself can be a bottleneck.
💻Code: github.com/shiv-ram-repo/con…
📜Paper: arxiv.org/abs/2606.08100
#ProteinStability #DDG #ProteinEngineering #ComputationalBiology #MachineLearning #FoundationModels #OODGeneralization #RepresentationLearning #ESM2 #ProteinMPNN
5
28
1,666
Generative pretraining for drug molecule design with bidirectional structure-property optimization
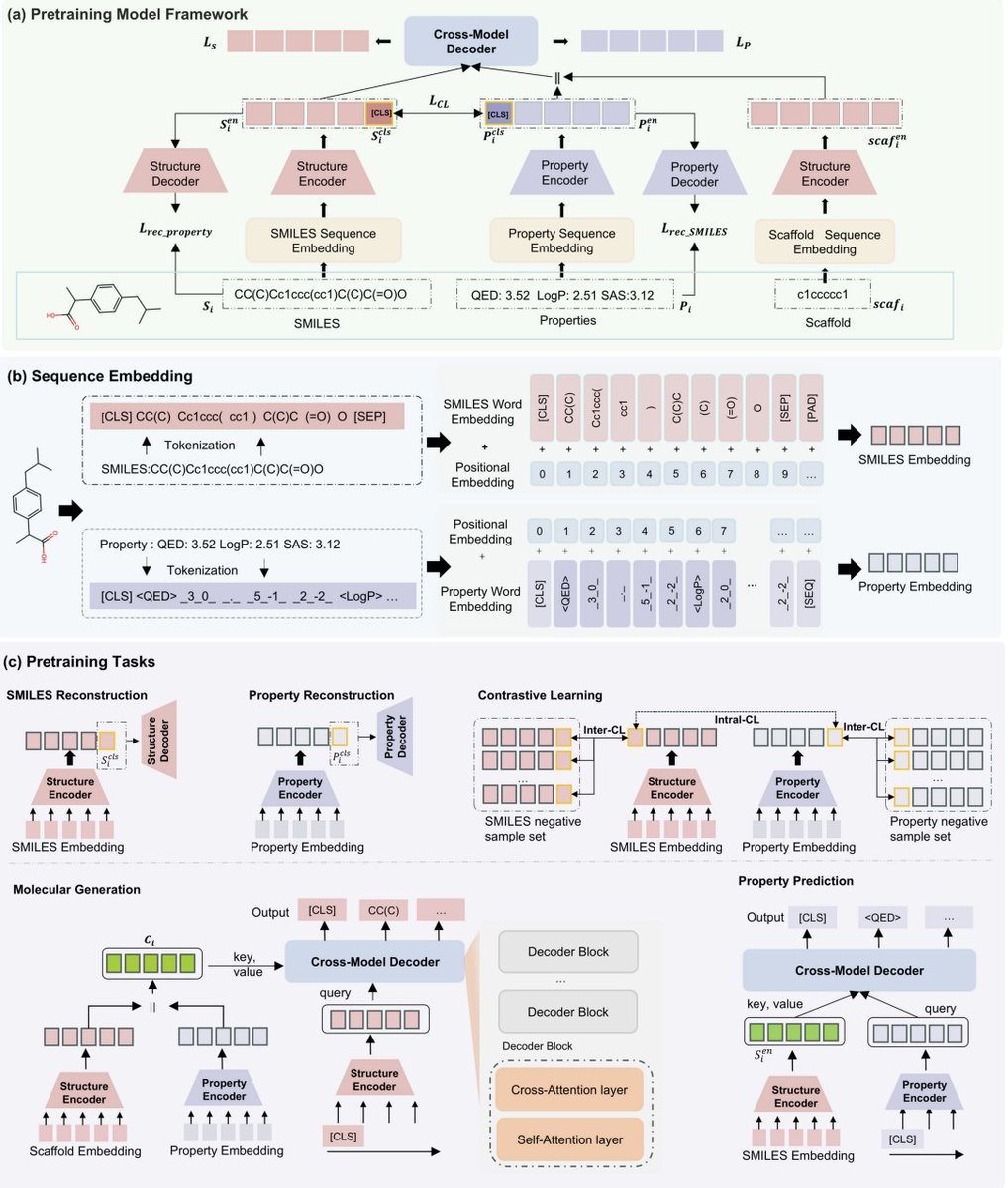
1. The paper presents BiSP-GP, a single pretrained framework that supports both controllable molecule generation (properties and/or scaffolds as conditions) and SMILES-to-property prediction, using one unified autoregressive sequence modeling setup rather than separate task-specific models.
2. A key idea is to turn continuous properties into “language”: QED, LogP, and SAS are serialized into semantic token sequences (property identifier, sign, digits, decimal point, and digit position tokens). This keeps numerical precision while letting properties be modeled in the same token space as SMILES, avoiding the usual “properties as plain numeric constraints” design.
3. Architecture: dual Transformer encoders (structure encoder for SMILES/scaffolds; property encoder for property-token sequences) plus a cross-modal decoder with cross-attention. The decoder enables bidirectional mapping: (a) generate SMILES conditioned on properties/scaffolds, and (b) generate property tokens conditioned on SMILES.
4. Pretraining uses five self-supervised objectives: SMILES reconstruction, property reconstruction, cross-modal intra-modal contrastive learning, conditional SMILES generation, and SMILES-conditioned property generation. The contrastive part includes a soft-label strategy (via momentum encoder) to reduce false negatives among structurally similar molecules with similar properties.
5. Robustness mechanism: stochastic masking of conditions. With 50% probability, an entire property’s tokens are replaced by [UNK], exposing the model to missing/incomplete property settings and enabling flexible inference-time control (choose which properties to constrain by providing tokens; leave others as [UNK]).
6. Unconditional generation (1,000 samples) is compared to CharRNN, LatentGAN, MolGPT, SPMM, and GP-MoLFormer. BiSP-GP reports the best composite V*U*N*I score (0.804) with strong validity (0.986), near-perfect uniqueness (0.999), high novelty (0.926), and high internal diversity (0.882), aiming for a better novelty–diversity balance than several baselines.
7. Single-property conditional generation (targets across QED, LogP, SAS) is evaluated with mean absolute deviation (MAD) for control accuracy plus Moses quality metrics. BiSP-GP shows the lowest MAD across all three properties versus CMGN, Scaffold-GGM, and SPMM, while maintaining strong uniqueness and internal diversity under constraints.
8. Multi-property control is tested for QED-LogP, QED-SAS, LogP-SAS, and QED-LogP-SAS conditions. The model maintains validity/uniqueness/novelty > 0.9 across scenarios and produces property distributions clustered around targets, while leaving unconstrained properties broadly distributed—useful for realistic multi-objective optimization.
9. Scaffold-conditioned and scaffold property generation: on 100 unseen scaffolds, BiSP-GP keeps scaffold similarity ratio (Sim_ratio) > 0.8 while generating novel variants; similarity analyses suggest novelty comes from both out-of-distribution scaffolds and side-chain diversification. Joint scaffold multi-property constraints still preserve scaffold structure with property values concentrated near targets.
10. Practical case study: PAK1 inhibitor optimization. With a fixed scaffold and a reduced LogP target (from 4.70 down toward 2.50 while holding QED and SAS), generated candidates show improved docking scores on PAK1 (PDB: 4EQC) on average (~0.35 kcal/mol better than the reference) and introduce additional polar interactions while retaining a key H-bond with GLU-315.
11. Property prediction as sequence generation: on 1,000 unseen molecules, BiSP-GP generates grammatically valid property strings and achieves very high agreement with RDKit-computed values (R²: LogP 0.999, QED 0.997, SAS 0.987). It remains reliable on randomized SMILES, suggesting learned structure–property relationships are not brittle to SMILES syntax variation.
12. Transfer learning: using the pretrained structure encoder as a frozen feature extractor plus a lightweight head, BiSP-GP performs strongly on MoleculeNet tasks plus Malaria and CEP, with statistically supported gains over several baselines on many regression/classification datasets; y-scrambling checks indicate performance is not driven by label artifacts.
13. Ablations indicate both innovations matter: replacing property serialization with numeric embeddings degrades conditional control (notably LogP MAD) and lowers property-prediction R²; removing contrastive learning broadly reduces generation quality, controllability, and prediction accuracy—supporting the role of cross-modal alignment.
💻Code: github.com/xmubiocode/BiSP-G… (Zenodo: zenodo.org/records/20115955)
📜Paper: doi.org/10.1038/s42004-026-0…
#ComputationalChemistry #Cheminformatics #MolecularGeneration #DrugDiscovery #Transformers #FoundationModels #GenerativeAI #PropertyPrediction #ScaffoldHopping #RepresentationLearning
2
23
1,588
When Does Structure Help? The Information Bonus of AlphaFold2 Representations over Protein Language Models
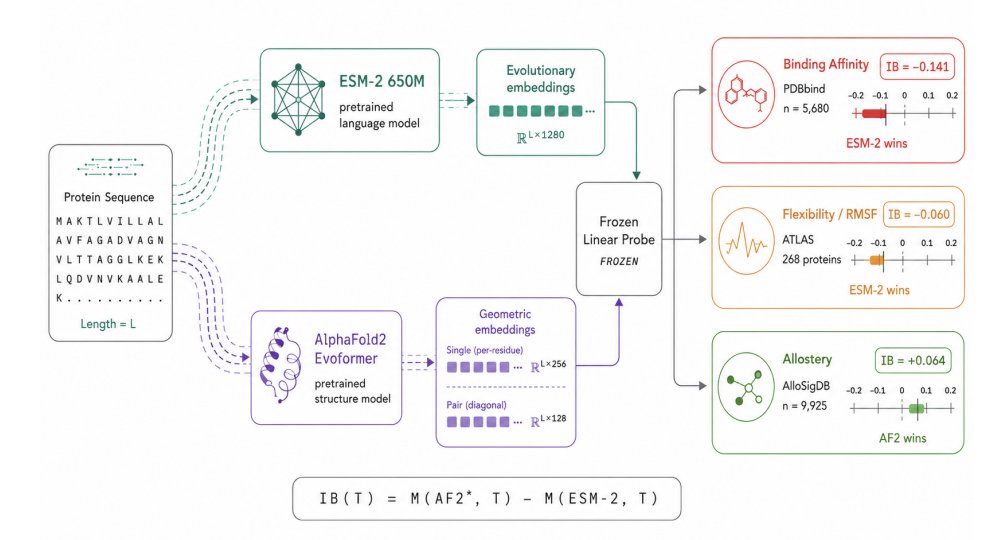
1. The paper introduces Information Bonus (IB): a task-level metric that quantifies how much linearly accessible signal is gained by using frozen AlphaFold2 (AF2) Evoformer representations instead of a cheaper frozen sequence-only model (ESM-2), evaluated under protein-level cross-validation.
2. IB is defined as the held-out performance difference between the best AF2 representation (chosen post-hoc between Evoformer single vs pair-diagonal) and ESM-2, using the same frozen linear probe. IB > 0 means structure adds usable signal; IB < 0 means sequence embeddings are sufficient or better.
3. The most decisive positive-IB regime is allostery (AlloSigDB; 47 proteins, 9,925 residues, 4.8% positives): AF2 single achieves AUROC 0.548, while ESM-2 is below chance at 0.485 and AF2 pair-diagonal is near chance at 0.497. This suggests AF2 single encodes long-range geometric/communication-network information that is not linearly recovered from sequence alone.
4. Binding affinity (PDBbind; n=5,680 complexes) shows a strong negative IB: ESM-2 reaches Pearson r=0.449 vs AF2 single r=0.307 and AF2 pair-diagonal r=0.278 (IB = -0.141). The paper argues this likely reflects evolutionary/family-level binding constraints captured by sequence models.
5. A key experimental design choice: the affinity probe receives only protein features (no ligand representation). So the benchmark tests whether representations capture protein-level correlates of affinity (e.g., pocket druggability, family propensity), not ligand-specific complementarity; AF2 features also reflect an apo-like inference rather than the bound complex.
6. Flexibility (ATLAS MD; 268 proteins, 50,426 residues) is mixed and label-dependent. For RMSF regression, AF2 pair-diagonal is directionally best (r=0.436) vs ESM-2 (r=0.407), giving a small positive IB ( 0.030) with limited statistical power across 5 folds.
7. For within-protein median flexibility classification, ESM-2 wins clearly: AUROC 0.824 vs AF2 pair-diagonal 0.764 and AF2 single 0.762 (IB = -0.060; p=0.0017 vs AF2 pair). Interpretation: sequence context captures disorder/mobility signatures better than static geometry for this relative-flexibility label.
8. The paper highlights a residue-level leakage artifact: naive residue-wise KFold (allowing residues from the same protein in both train/test) inflates RMSF performance by 27–39% depending on representation (e.g., ESM-2 r=0.672 under leaky split vs 0.407 under protein-level GroupKFold). This inflation can reverse representation rankings and change conclusions.
9. Practical takeaway framed for AI-scientist workflows: representation choice should be a measurable decision. Start with ESM-2 when labels are plausibly driven by evolutionary constraints or disorder-like sequence signatures; pay the AF2 inference cost when the mechanism depends on long-range 3D communication (as in allostery). When uncertain, estimate IB on a small labeled set before scaling structural inference.
📜Paper: arxiv.org/abs/2606.04228
#ComputationalBiology #ProteinML #AlphaFold2 #ProteinLanguageModels #RepresentationLearning #Allostery #Benchmarking #DataLeakage #AIFORScience
5
32
2,269

Jun 4
What information is actually hidden inside a multimodal embedding?
In this new work, we find that frozen vision-language models already encode rich attribute-specific signals for objects, backgrounds, and styles, even though their standard embeddings appear highly entangled.
We introduce QARE (Queryable Attribute Representation Extraction), a simple text-guided framework that extracts attribute-specific representations from frozen VLMs without fine-tuning.
Along the way, we build QARE-Bench, a challenging benchmark with both controlled synthetic data and a new real-world dataset featuring diverse scenes, non-rigid objects, and hard negatives designed to stress-test attribute disentanglement.
Key finding:
👉 The problem may not be that VLMs lack disentangled representations.
👉 The problem may be that we haven't learned how to query them.
📄 Paper: openaccess.thecvf.com/conten…
💻 Code: github.com/yibingwei-1/QARE
#ComputerVision #MultimodalAI #VisionLanguageModels #RepresentationLearning #ImageRetrieval

6
19
2,691

Maybe the data-efficiency gap is not a scaling problem.
Maybe it is an objective problem.
A striking preprint by Daniel J. Korchinski, Alessandro Favero, and Matthieu Wyart offers a sample-complexity theory for this shift:
Learn from your own latents and not from tokens.
The core problem is familiar:
modern generative models are extraordinary, but brutally data-hungry.
LLMs train on 10¹³–10¹⁴ tokens.
Children do not.
So the question is not only:
How do we scale models?
It is:
What are we asking them to predict?
Most of modern AI trains on the visible surface: next tokens, masked tokens, pixels, noise.
That works.
But it may be statistically inefficient for learning hierarchy.
The authors study a tractable hierarchical grammar where visible tokens are generated from a hidden latent tree of depth L — a stylized model for the compositional structure of language and images.
The result reframes the debate:
token-level learning requires samples exponential in L to recover the hidden tree.
latent prediction recovers it with sample complexity essentially constant in L, up to logarithmic factors.
In plain English:
predicting tokens forces the model to infer the hierarchy through the leaves.
predicting latents lets the model climb the tree.
Once one abstraction level is learned, it becomes the substrate for learning the next.
This is why data2vec and JEPA-style objectives are so interesting.
They do not merely reconstruct the input.
They train a network to predict its own latent representation of another view or masked region.
The target is no longer the surface.
The target is the model’s own emerging abstraction.
The paper validates the theory three ways:
a hierarchical clustering algorithm
an end-to-end neural architecture trained by gradient descent
a sample-complexity analysis of data2vec, showing it implicitly performs hierarchical latent prediction
One implication is provocative:
if data2vec already discovers hierarchy implicitly, explicit stacking schemes such as H-JEPA may be partly redundant.
This is not “next-token prediction is dead.”
Next-token prediction built the current era.
But if the goal is biological-level data efficiency, surface reconstruction may be the expensive path.
The strategic frontier may be latent self-prediction:
models learning not only from what they see,
but from the abstractions they are forming.
Full credit to the authors:
Daniel J. Korchinski, Alessandro Favero, Matthieu Wyart.
Paper:
Learn from your own latents and not from tokens: A sample-complexity theory
arxiv.org/abs/2605.27734
I’m attaching the first page because the abstract is worth reading closely.
The future of data-efficient AI may not be more tokens.
It may be better targets.
#AIResearch #MachineLearning #SelfSupervisedLearning #RepresentationLearning #DataEfficiency #LLM

3
1
10
1,024

May 31
The data-efficiency gap between machines and children may not be solved by “more tokens.”
It may be solved by changing what the model is asked to predict.
A beautiful new paper by Daniel J. Korchinski, Alessandro Favero, and Matthieu Wyart gives a sample-complexity theory for a major alternative to token-level learning:
Learn from your own latents and not from tokens.
The premise is striking.
Modern generative models learn by predicting raw surface fragments:
next tokens
masked tokens
pixels
noise
patches
This works spectacularly.
But it is brutally data-hungry.
Biological learners do not see 10¹³–10¹⁴ tokens before acquiring rich language competence. So perhaps the bottleneck is not only architecture, scale, or optimization.
Perhaps it is the prediction target.
Instead of predicting tokens, methods like data2vec and JEPA train networks to predict their own latent representations of related views or masked regions.
The model is not asked:
“Can you reconstruct the surface?”
It is asked:
“Can you predict the abstraction your own system would form?”
That difference may be enormous.
The authors study a tractable hierarchical grammar that generates visible tokens from hidden latent trees of depth L — a stylized model of the compositional structure of language and images.
For this data, supervised learning and token-level self-supervised learning require samples exponential in L to recover the hidden hierarchy.
But latent prediction recovers the hierarchy with sample complexity essentially constant in L, up to logarithmic factors.
That is the whole paper in one line:
predicting tokens makes hierarchy expensive;
predicting latents makes hierarchy recursive.
Why?
Because token-level objectives keep forcing supervision through the visible surface. The deeper the hidden structure, the weaker and more indirect the signal becomes.
Latent prediction removes that bottleneck.
Once one level of abstraction is recovered, the model can use its own learned latents as both context and target for the next level. Every level becomes statistically like the first.
The paper confirms this three ways:
a hierarchical clustering algorithm
an end-to-end neural architecture trained by gradient descent
a sample-complexity analysis of data2vec, showing that it implicitly performs hierarchical latent prediction
The last point is especially interesting.
If data2vec already discovers hierarchy implicitly, then explicitly stacking methods like H-JEPA may be partly redundant.
This is not “tokens are dead.”
Token prediction remains one of the most productive ideas in AI.
But this paper gives a precise reason why token-level learning may be an inefficient path to latent structure.
The deeper lesson:
the model should not only learn from the world’s surface.
It should learn from the abstractions it is already beginning to form.
Full credit to the authors:
Daniel J. Korchinski, Alessandro Favero, Matthieu Wyart.
Paper:
Learn from your own latents and not from tokens: A sample-complexity theory
arxiv.org/abs/2605.27734
I’m attaching the first page because the abstract is worth reading closely.
The future of data-efficient AI may not be more reconstruction.
It may be recursive self-prediction in latent space.
#AIResearch #MachineLearning #SelfSupervisedLearning #RepresentationLearning #LLM #DataEfficiency

1
3
6
350
Fragmentnet: Adaptive graph fragmentation for graph-to-sequence molecular representation learning
1. FragmentNet reframes molecular pretraining around learned, chemically valid fragments (not atoms): it serializes a molecular graph into a fragment sequence, masks an entire fragment, and trains a Transformer to reconstruct that missing substructure (Masked Fragment Modeling, MFM).
2. The core technical piece is an adaptive graph tokenizer that starts from atoms and iteratively merges connected pairs using a corpus-driven score (pair frequency normalized by node frequencies), storing a merge history so granularity can be changed at inference/fine-tune time by choosing how many merges to apply.
3. Unlike rigid rule-based fragmentation (e.g., BRICS) or SMILES subword tokenization, the tokenizer preserves graph connectivity and chemical validity, and explicitly represents cut points via dummy atoms (atomic number 0), so “the same” fragment with different dangling-bond environments becomes distinct tokens.
4. To uniquely index fragments (including stereochemical variants and dangling-bond context), FragmentNet builds its token dictionary using Weisfeiler–Lehman (WL) hashing with atom labels (Z, hybridization, radicals, H count) and bond labels (type, conjugation, stereo, ring membership), avoiding SMILES non-uniqueness.
5. The model is a hybrid graph-to-sequence pipeline: a VQ-VAE encodes discrete atom-level attributes into codebooks, a GCN captures intra-fragment structure, and the two are combined into fragment embeddings that are then processed by a BERT-style Transformer.
6. A key challenge in graph-to-sequence is preserving topology after serialization; FragmentNet adds “chemically aware” spatial positional encodings by summing (i) hop-based global distance summaries, (ii) WL absolute/role encodings, and (iii) Coulomb-matrix-inspired charge/interaction encodings.
7. It also replaces the standard CLS token with a learnable molecular-descriptor vector (computed from RDKit descriptors and refined through attention), aiming to provide a global summary channel alongside fragment-context modeling.
8. Empirically, with MFM pretraining on 2M molecules, fragment-level tokenization (100 merges; ~7 fragments per molecule, ~10 atoms per token on average) beats atom-level tokenization (0 merges) on 5/7 scaffold-split benchmarks (MoleculeNet Malaria); without pretraining, atom-level often does better, highlighting that granularity interacts strongly with pretraining.
9. Beyond prediction, the learned fragment vocabulary enables a fragment-swapping module for targeted analogue generation: by matching dummy-atom bond environments and sanitizing with RDKit, it can substitute fragments while preserving the core scaffold (demonstrated on ibuprofen, aspirin, diazepam) without expensive substructure search.
📜Paper: arxiv.org/abs/2502.01184
#ComputationalChemistry #Cheminformatics #MolecularML #GraphML #Transformers #SelfSupervisedLearning #DrugDiscovery #RepresentationLearning
3
940

May 27
🎉 Excited to share our new work accepted to #CVPR2026 “𝗡𝗲𝘅𝘂𝘀𝗙𝗹𝗼𝘄: 𝗨𝗻𝗶𝗳𝘆𝗶𝗻𝗴 𝗗𝗶𝘀𝗽𝗮𝗿𝗮𝘁𝗲 𝗧𝗮𝘀𝗸𝘀 𝘂𝗻𝗱𝗲𝗿 𝗣𝗮𝗿𝘁𝗶𝗮𝗹 𝗦𝘂𝗽𝗲𝗿𝘃𝗶𝘀𝗶𝗼𝗻 𝘃𝗶𝗮 𝗜𝗻𝘃𝗲𝗿𝘁𝗶𝗯𝗹𝗲 𝗙𝗹𝗼𝘄 𝗡𝗲𝘁𝘄𝗼𝗿𝗸𝘀”
In textbooks and benchmarks, datasets are often neatly annotated for every task. In the real world, they rarely are. Data is collected at different times, in different places, and for different purposes. One dataset may contain labels for mapping, another for tracking, another for depth or segmentation. Does that mean fragmented data has to be discarded?
💪 𝗢𝘂𝗿 𝗮𝗻𝘀𝘄𝗲𝗿: 𝗻𝗼. We show that partially supervised, heterogeneous data can still be highly valuable—and in some cases, can even outperform fully annotated data.
How do we learn across structurally different tasks when labels are only partially available?
💡 𝗢𝘂𝗿 𝗦𝗼𝗹𝘂𝘁𝗶𝗼𝗻: 𝗡𝗲𝘅𝘂𝘀𝗙𝗹𝗼𝘄
NexusFlow is a lightweight, plug-and-play framework that aligns disparate tasks in a shared latent space.
What makes it work:
• 🔄 𝗜𝗻𝘃𝗲𝗿𝘁𝗶𝗯𝗹𝗲 𝗳𝗲𝗮𝘁𝘂𝗿𝗲 𝗮𝗹𝗶𝗴𝗻𝗺𝗲𝗻𝘁. Invertible coupling layers map task features into a unified canonical space. Since the mapping is bijective, task information is preserved, helping avoid the representational collapse often seen in vanilla alignment methods.
• 🔌 𝗣𝗹𝘂𝗴-𝗮𝗻𝗱-𝗽𝗹𝗮𝘆 𝗱𝗲𝘀𝗶𝗴𝗻. No need to modify task heads or losses. NexusFlow can be added to BEV-based backbones with a simple alignment loss.
• 📈 𝗦𝗰𝗮𝗹𝗮𝗯𝗹𝗲 𝘁𝗼 𝗺𝘂𝗹𝘁𝗶𝗽𝗹𝗲 𝘁𝗮𝘀𝗸𝘀. The method scales as O(N) with one surrogate branch per task, making extension to 3 tasks straightforward.
• 📐 𝗧𝗵𝗲𝗼𝗿𝗲𝘁𝗶𝗰𝗮𝗹 𝗴𝗿𝗼𝘂𝗻𝗱𝗶𝗻𝗴. Invertibility provides a provable bound that connects the alignment loss to cross-task knowledge transfer.
🏆 𝗥𝗲𝘀𝘂𝗹𝘁𝘀
NexusFlow sets a new state of the art on nuScenes for domain-partitioned autonomous driving, where online map reconstruction and multi-object tracking are supervised in different geographic regions.
It also delivers consistent gains across all three NYUv2 tasks: semantic segmentation, depth estimation, and surface normal prediction.
📎 𝗣𝗿𝗼𝗷𝗲𝗰𝘁 𝗽𝗮𝗴𝗲: ark1234.github.io/nexusflow_…
🤝 This work was conducted in collaboration across Worcester Polytechnic Institute, Texas A&M University, Tohoku University, University of Michigan, and Bosch Research.
Huge thanks to collaborators: Fangzhou Lin, Yuping Wang, Yuliang Guo, Zixun Huang, Xinyu Huang, Haichong Zhang, Kazunori Yamada, Zhengzhong Tu, Liu Ren, and Ziming Zhang.
#CVPR2026 #ComputerVision #MultiTaskLearning #AI #GenAI #AutonomousDriving #DeepLearning #RepresentationLearning
1
18
2,130
May 27
Language models may not need to “build” hierarchies.
Hierarchies may fall out of the statistics of language.
A beautiful new paper by Andres Nava and Matthieu Wyart proposes a distributional theory for one of the most basic structures in meaning:
the “is-a” relation.
An owl is a bird.
A bird is an animal.
An animal is an organism.
This relation — hypernymy — looks like an ontology.
But the paper asks a sharper question:
Does hierarchical concept geometry in language models require a hierarchy-specific mechanism?
Or can it emerge from word co-occurrence alone?
Their answer is striking.
Start with a simple empirical fact:
words closer together in the WordNet hierarchy tend to co-occur more often.
“tree” and “plant” appear together more than “tree” and “organism.”
That decay in co-occurrence with semantic distance induces structure in the embedding Gram matrix.
Then the spectrum does the rest.
The leading eigenvectors first separate broad branches of the taxonomy, then progressively finer sub-branches.
This creates what the authors call hierarchical splitting geometry:
coarse-to-fine organization in representation space.
In the organism example, one principal direction separates plants from animals. Later directions split flowers from trees, birds from fish, and eventually finer distinctions like daisy vs. poppy.
That is the elegant part:
the geometry looks conceptual,
but the mechanism is spectral.
The authors prove this under mild positivity and decay assumptions on the co-occurrence kernel, confirm it across sampled WordNet subtrees in word2vec, and then show the same signature extends surprisingly well to Gemma 2B unembeddings.
This is not saying LLMs do not represent hierarchies.
They clearly do.
It is saying we should be careful about why that geometry exists.
Some elegant semantic structure may not be evidence of a specialized internal ontology.
It may be the mathematical shadow of pairwise word statistics.
That matters for interpretability.
If we find clean concept directions, orthogonal refinements, or taxonomic splits inside models, we should ask:
Is this a functional mechanism?
Or is it the spectrum of the data distribution made visible?
This paper pushes toward a more precise science of representation geometry.
Less mysticism.
More mechanism.
Less “the model learned an ontology.”
More “the co-occurrence kernel shaped an eigenspace.”
Full credit to the authors:
Andres Nava and Matthieu Wyart.
Paper:
Hierarchical Concept Geometry in Language Models Emerges from Word Co-occurrence
arxiv.org/abs/2605.23821
I’m attaching the first page because Figure 1 is worth studying closely.
The deep lesson:
meaning may become geometry not because the model was taught a taxonomy,
but because language itself already contains one in its statistics.
#AIResearch #Interpretability #LLM #NLP #RepresentationLearning #MachineLearning

9
42
197
11,069
What looks like ontology may be eigenspectrum.
A beautiful new paper by Andres Nava and Matthieu Wyart gives a mechanistic account of one of the most striking facts about language models:
semantic hierarchies appear geometrically.
An owl is a bird.
A bird is an animal.
An animal is an organism.
In representation space, these “is-a” relations often seem to organize into clean directions, subspaces, and taxonomic refinements.
The tempting interpretation is functional:
the model learned an internal ontology.
This paper asks a more dangerous question:
what if part of that geometry is not an engineered semantic mechanism at all?
What if it is the spectral shadow of word co-occurrence?
The core move is elegant.
Start with WordNet.
Measure semantic distance in the hypernym graph.
Verify that closer concepts co-occur more often.
Then analyze the Gram matrix induced by those pairwise word statistics.
Under mild positivity and decay assumptions, the leading eigenvectors separate the taxonomy from coarse to fine.
First, broad branches split.
Plant vs. animal.
Then finer branches split.
Flower vs. tree.
Bird vs. fish.
Daisy vs. poppy.
This is what the authors call hierarchical splitting geometry.
The remarkable part is that the same structure appears in simple word2vec embeddings and extends strikingly well to Gemma 2B unembeddings.
That matters.
Because it suggests that some concept geometry in LLMs may not require a hierarchy-specific module, circuit, or functional objective.
It can emerge from the spectrum of pairwise language statistics.
In other words:
language already contains a tree,
co-occurrence encodes distances on that tree,
and spectral decomposition turns those distances into geometry.
This is a serious interpretability lesson.
When we find clean semantic directions inside a model, we should not immediately ask:
“What internal mechanism built this ontology?”
We should also ask:
“What structure in the data distribution made this geometry inevitable?”
That distinction is crucial.
Functional geometry asks what a representation can do.
Distributional geometry asks where the representation came from.
This paper pushes interpretability toward a more mature science:
less anthropomorphic storytelling,
more spectral mechanism.
Less “the model has a taxonomy in its head,”
more “the co-occurrence kernel shaped an eigenspace.”
Full credit to the authors:
Andres Nava and Matthieu Wyart.
Paper:
Hierarchical Concept Geometry in Language Models Emerges from Word Co-occurrence
arxiv.org/abs/2605.23821
I’m attaching the first page because Figure 1 is worth studying closely.
The deep lesson:
meaning may become geometry not because the model was explicitly taught hierarchy,
but because language statistics already carry one.
#AIResearch #Interpretability #LLM #NLP #RepresentationLearning #MachineLearning

3
13
74
4,653

⚠️ Limited seats remaining for MLx Representation Learning & Generative AI at Oxford Maths Institute Online (15–18 July).
Join leading researchers and practitioners exploring frontier models, scaling laws, modern architectures, generative AI systems, representation learning, and AI products.
Some of the Featured Lectures:
Why formalize mathematics— Kevin Buzzard (Imperial College London)
Intelligent Data Gathering— Tom Rainforth (University of Oxford)
Multi-Robot and Multi-Agent Learning— Amanda Prorok (University of Cambridge)
Embodied Multimodal Intelligence with Foundation Models— Oier Mees (Microsoft)
Multimodal AI— Paul Liang (MIT)
On Causal Discovery and the Extrapolation of Causal Effects— Ricardo Silva (UCL)
A theoretical view with Arena's data—Peter Gostev (Arena AI)
Petar Veličković (Google DeepMind)
Fazl Barez (University of Oxford)
Tim Rocktäschel (UCL)
Alexander Tong (Aithyra)
Tony Feng UC Berkeley
Register now before seats fill up.
oxfordml.school
@FazlBarez @AlexanderTong7 @_rockt @petergostev @pliang279 @oier_mees @aprorok @tom_rainforth
#MachineLearning #GenerativeAI #RepresentationLearning #AI #OxML

4
11
928

May 17
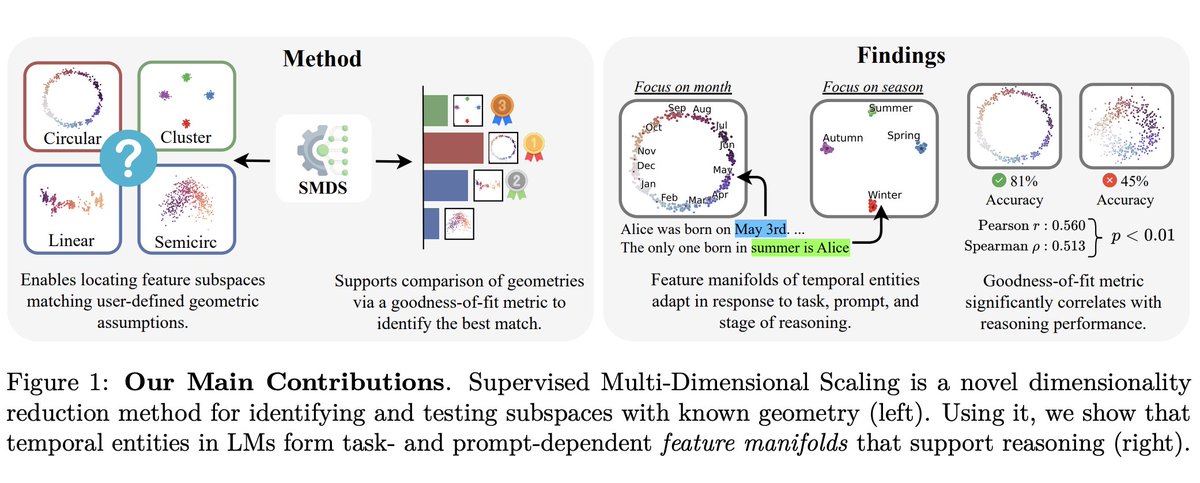
A really interesting paper on representation geometry in LLMs written by my friend @frankniujc :
“Hypothesis-Driven Feature Manifold Analysis in LLMs via SMDS” proposes a model-agnostic way to test geometric hypotheses about latent representations instead of assuming everything is just linear directions. They find that different concepts naturally form different structures like circles, lines, clusters, and that these manifolds remain surprisingly stable across model families/sizes while also dynamically reshaping with context. Very cool bridge between mechanistic interpretability and representation geometry. 🔥
Especially liked the framing that reasoning may operate over structured manifolds rather than isolated features.
Paper: openreview.net/pdf?id=vCKZ40…
Code: github.com/UKPLab/tmlr2026-m…
#LLM #MechanisticInterpretability #AIResearch #RepresentationLearning #TMLR #Interpretability #DeepLearning



6
36
219
21,572
Unified Genomic and Chemical Representations Enable Bidirectional Biosynthetic Gene Cluster and Natural Product Retrieval
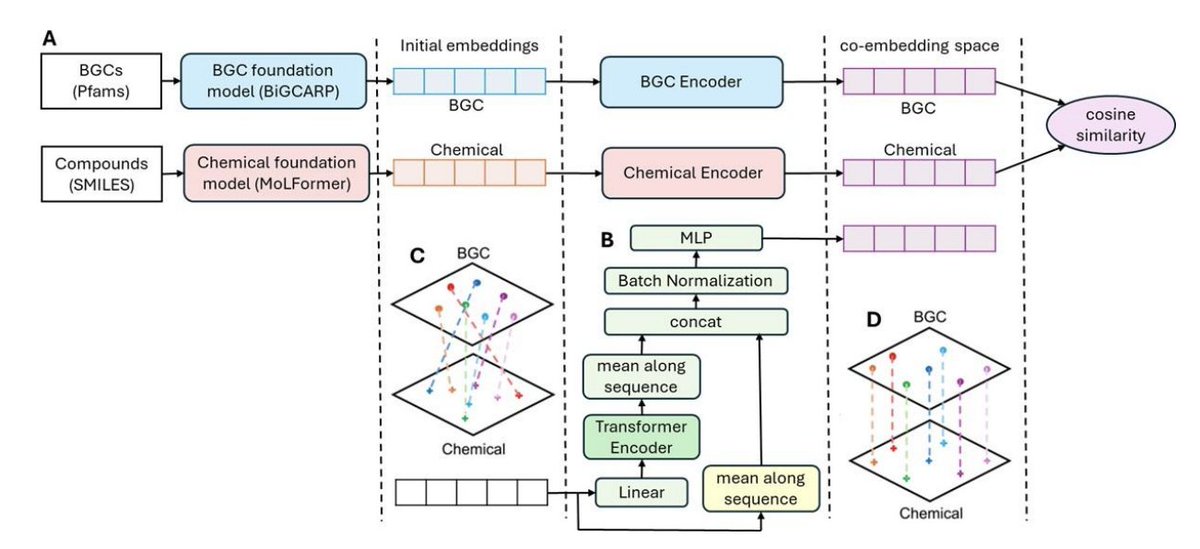
1. Liu, Li, Ong et al. present BCCoE, a multimodal retrieval framework that puts biosynthetic gene clusters (BGCs) and natural products into a shared embedding space, enabling both directions of search: BGC→compound and compound→BGC.
2. The key idea is to reuse strong pretrained “foundation” embeddings from each modality, then learn only a lightweight alignment: BiGCARP embeddings for BGC Pfam-domain sequences (256D) MoLFormer embeddings for compound SMILES (768D), projected into a 64D co-embedding space for cosine-similarity nearest-neighbor retrieval.
3. Architecture: two modality-specific encoders (same structure, separate weights) that apply (i) linear projection, (ii) a 2-layer transformer encoder, (iii) pooling concatenation with the mean of the original embedding sequence, then (iv) batch norm a 2-layer MLP to output the final co-embedding vectors.
4. Training is metric learning with N-pair loss over batches of paired (BGC, compound) examples from MIBiG; foundation-model embeddings are frozen to reduce overfitting and to preserve general representations. Negatives are implicitly taken from other pairs within the same batch (efficient “in-batch” negatives).
5. Why alignment matters: baselines that do retrieval without cross-modal alignment (KNN and a two-hop KNN-2hop that chains BGC-similarity and compound-similarity) cannot consistently capture genotype–chemotype links, especially when candidate pools include novel items not seen during training.
6. Main quantitative results on MIBiG 4.0 (10-fold CV): for BGC→compound retrieval at top-10, Recall improves from 12.9% (KNN) and 21.9% (KNN-2hop) to 32.9% (BCCoE); for compound→BGC at top-10, BCCoE reaches 65.3% Recall (vs 60.6% KNN-2hop), with very large lift over random guessing at low K.
7. Generalization to unseen product classes (hold out one entire BGC product class during training): performance drops for all methods, but BCCoE remains substantially better, achieving Lift@10 of 17.0 (BGC→compound) and 20.2 (compound→BGC), outperforming KNN-2hop by ~75–89% in lift at top-10.
8. Temporal generalization (train on MIBiG 3.1, evaluate on new links added in MIBiG 4.0): BCCoE improves identification of newly added BGC–compound pairs, e.g., when retrieving compounds from the full MIBiG 4.0 candidate set, top-10 hits rise from 126 (KNN-2hop) to 180 (BCCoE) among 473 new pairs.
9. Robustness across alternative foundation models: swapping in ESM-C for BGCs or Uni-Mol2 for compounds shows BCCoE remains relatively stable, while KNN-2hop can degrade sharply due to “similarity saturation” (cosine similarities clustered near 1 in the initial embedding spaces), which breaks two-hop score ranking; BCCoE’s aligned space yields a more well-behaved similarity distribution.
10. Practical validation beyond MIBiG: on three experimentally validated external BGC–compound pairs previously used in BGC-MAP, BCCoE ranks the true matches much higher in both directions (BGC→compound and compound→BGC), supporting its use for prioritizing candidates in real discovery workflows.
💻Code: zenodo.org/records/18849052
📜Paper: doi.org/10.1038/s41598-026-4…
#Bioinformatics #ComputationalBiology #NaturalProducts #GenomeMining #BiosyntheticGeneClusters #MultimodalAI #MetricLearning #RepresentationLearning #Cheminformatics
3
14
2,091

🤗 Hugging Face Model (PyTorch): huggingface.co/InstaDeepAI/M…
🧑🏻💻 Repository (JAX): github.com/instadeepai/multi…
See you at ICML! 🇰🇷
#ICML #ICML2026 #MachineLearning #MultiOmics #CancerResearch #RepresentationLearning #RNAseq #DNAmethylation #HuggingFace #JAX #PyTorch #MOJO #InstaDeep
3
100
Graph neural network based hierarchy-aware embeddings of knowledge graphs: Applications to yeast phenotype prediction
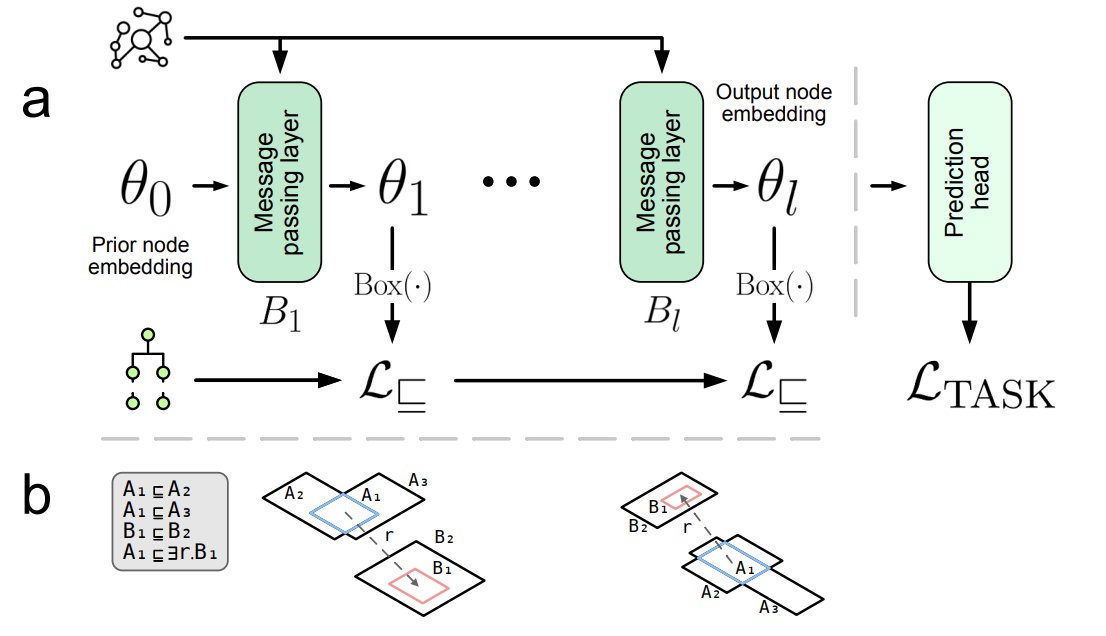
1. Kronström et al. introduce Hierarchy-aware GNNs: a framework that couples GNN message passing on heterogeneous KGs with box embeddings constrained by ontology class hierarchies via a semantic loss, so learned representations better respect domain semantics.
2. Key idea: treat each GNN layer’s node output as latent variables that parameterise axis-aligned boxes; then enforce ontology constraints (e.g., subClassOf as geometric containment, and optional disjointness as non-overlap) using semantic losses during end-to-end training.
3. The KG is built for Saccharomyces cerevisiae by integrating curated resources (SGD, GO, APO, ChEBI, INO, MI, RO, BioCyc) and rewriting ABox facts into a TBox-style form, enabling a uniform representation of individuals/classes and relations as axioms usable for both GNN computation and semantic constraints.
4. The model targets quantitative phenotype prediction: predicting fitness (cell growth) for double gene deletions (digenic knockouts) as an edge-level regression problem over gene nodes, trained on 10,085,183 gene-pair measurements (standard condition 30°C), with careful removal of overlapping interaction edges from the KG to avoid leakage.
5. Architecture details: a heterogeneous GraphSAGE-style GNN (relation-specific modules per source-edge-target type) produces gene embeddings; deleted gene pairs are combined with a symmetric operator (best: element-wise product) and passed to an MLP regressor. Domains are embedded separately (8 ontology-aligned domains) to improve stability and efficiency.
6. Main performance result (10-fold CV, split by genes to prevent overlap between train/validation genes): a task-only GNN reaches mean R2=0.348; adding subClassOf links directly to the KG yields R2=0.350; using pretrained box embeddings as priors improves to R2=0.360; adding semantic loss improves further to R2=0.368 (overlap loss) and best R2=0.377 (distance-based loss).
7. Baselines: LightGBM on sparse phenotype instantiations achieves R2=0.211; LightGBM on a 64-dim ComplEx KG embedding achieves R2=0.191. This supports the claim that end-to-end heterogeneous GNN embeddings, especially with hierarchy-aware constraints, extract more predictive signal from the KG than these feature/embedding baselines.
8. Generalisation test: models trained on digenic deletions are evaluated on trigenic deletion fitness (15,095 triples). Using a 3-way element-wise product, performance reaches R2=0.380 with box priors, and R2=0.415 with distance-based semantic loss, indicating transfer beyond the original pairwise setting.
9. Interpretability-to-experiment loop: using gradient-based attribution over KG-linked traits, they score co-occurring relations important for predictions, generating hypotheses about interacting phenotypes. A selected, lab-feasible hypothesis linked inositol utilisation with NaCl (osmotic) stress resistance; an automated-lab perturbation experiment found a significant interaction, with inositol supplementation rescuing growth under salt stress.
10. Additional contributions: (i) shows how to learn low-dimensional GNN-driven box embeddings without a prediction task (pure semantic training), comparing distance vs overlap losses and visualising 2D molecular-function embeddings; (ii) explores using embedding changes under proposed edge additions as a way to rank KG revisions, finding some relation types show distinguishable rank distributions versus random edges, though effects can be small and variable.
📜Paper: arxiv.org/abs/2605.03690
#KnowledgeGraphs #GraphNeuralNetworks #Ontology #RepresentationLearning #BoxEmbeddings #ComputationalBiology #SystemsBiology #Yeast #PhenotypePrediction #ExplainableAI
5
1,172
Graph neural network based hierarchy-aware embeddings of knowledge graphs: Applications to yeast phenotype prediction
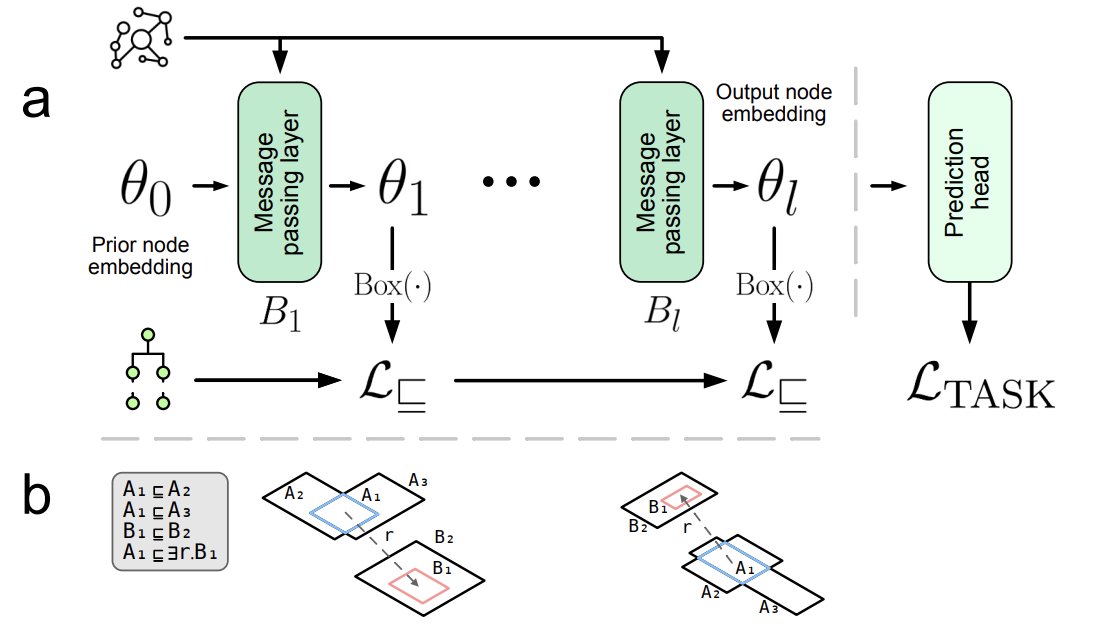
1. Kronström et al. introduce Hierarchy-aware GNNs: a framework that couples GNN message passing on heterogeneous KGs with box embeddings constrained by ontology class hierarchies via a semantic loss, so learned representations better respect domain semantics.
2. Key idea: treat each GNN layer’s node output as latent variables that parameterise axis-aligned boxes; then enforce ontology constraints (e.g., subClassOf as geometric containment, and optional disjointness as non-overlap) using semantic losses during end-to-end training.
3. The KG is built for Saccharomyces cerevisiae by integrating curated resources (SGD, GO, APO, ChEBI, INO, MI, RO, BioCyc) and rewriting ABox facts into a TBox-style form, enabling a uniform representation of individuals/classes and relations as axioms usable for both GNN computation and semantic constraints.
4. The model targets quantitative phenotype prediction: predicting fitness (cell growth) for double gene deletions (digenic knockouts) as an edge-level regression problem over gene nodes, trained on 10,085,183 gene-pair measurements (standard condition 30°C), with careful removal of overlapping interaction edges from the KG to avoid leakage.
5. Architecture details: a heterogeneous GraphSAGE-style GNN (relation-specific modules per source-edge-target type) produces gene embeddings; deleted gene pairs are combined with a symmetric operator (best: element-wise product) and passed to an MLP regressor. Domains are embedded separately (8 ontology-aligned domains) to improve stability and efficiency.
6. Main performance result (10-fold CV, split by genes to prevent overlap between train/validation genes): a task-only GNN reaches mean R2=0.348; adding subClassOf links directly to the KG yields R2=0.350; using pretrained box embeddings as priors improves to R2=0.360; adding semantic loss improves further to R2=0.368 (overlap loss) and best R2=0.377 (distance-based loss).
7. Baselines: LightGBM on sparse phenotype instantiations achieves R2=0.211; LightGBM on a 64-dim ComplEx KG embedding achieves R2=0.191. This supports the claim that end-to-end heterogeneous GNN embeddings, especially with hierarchy-aware constraints, extract more predictive signal from the KG than these feature/embedding baselines.
8. Generalisation test: models trained on digenic deletions are evaluated on trigenic deletion fitness (15,095 triples). Using a 3-way element-wise product, performance reaches R2=0.380 with box priors, and R2=0.415 with distance-based semantic loss, indicating transfer beyond the original pairwise setting.
9. Interpretability-to-experiment loop: using gradient-based attribution over KG-linked traits, they score co-occurring relations important for predictions, generating hypotheses about interacting phenotypes. A selected, lab-feasible hypothesis linked inositol utilisation with NaCl (osmotic) stress resistance; an automated-lab perturbation experiment found a significant interaction, with inositol supplementation rescuing growth under salt stress.
10. Additional contributions: (i) shows how to learn low-dimensional GNN-driven box embeddings without a prediction task (pure semantic training), comparing distance vs overlap losses and visualising 2D molecular-function embeddings; (ii) explores using embedding changes under proposed edge additions as a way to rank KG revisions, finding some relation types show distinguishable rank distributions versus random edges, though effects can be small and variable.
📜Paper: arxiv.org/abs/2605.03690
#KnowledgeGraphs #GraphNeuralNetworks #Ontology #RepresentationLearning #BoxEmbeddings #ComputationalBiology #SystemsBiology #Yeast #PhenotypePrediction #ExplainableAI
5
12
1,721
Aligning LLMs with Biomedical Knowledge using Balanced Fine-Tuning
1. The paper argues that “low-confidence tokens” mean something different in biomedicine: they often form dense contiguous runs that encode rare entities (genes/mutations/pathway nodes) and mechanistic causal chains—i.e., epistemic uncertainty (knowledge gaps)—rather than the sparse stylistic alternatives (aleatoric uncertainty) common in general text.
2. This observation motivates Balanced Fine-Tuning (BFT), a dual-scale post-training objective designed to keep learning signal on knowledge-dense uncertainty while still stabilizing optimization—addressing a key failure mode where Dynamic Fine-Tuning (DFT) down-weights exactly the biomedical tokens that matter.
3. The authors operationalize “dense epistemic uncertainty” with a teacher-forcing diagnostic: compute per-token confidence, slide a 256-token window, and classify windows by (a) fraction of low-confidence tokens and (b) longest contiguous low-confidence run. Sparse-low windows (Group A) tend to be stylistic; dense-low windows (Group B) are enriched for biomedical entities and causal connectives.
4. BFT token-level innovation: replace DFT’s absolute confidence weighting with group-normalized reweighting using a local context confidence (mean confidence in a g=256 sliding window). Each token weight is proportional to cb,t / (Clocb,t ε), clipped to [0,1] and stop-gradient detached—suppressing isolated low-confidence outliers while preserving gradients in globally hard (dense-low) biomedical spans.
5. BFT sample-level innovation: reallocate learning across sequences using a bounded hard-sample coefficient derived from the minimum local context confidence within the sequence. This explicitly shifts optimization budget toward samples containing the hardest knowledge-dense regions, complementing token-level gating.
6. Across tasks (medical evaluation, biological reasoning, sparse-reward RL, and representation learning), BFT provides more consistent gains than SFT and DFT under the same training recipe and model family (DeepSeek-R1-Distill 14B/32B/70B), suggesting the uncertainty-aware loss design transfers across biomedical settings.
7. Backbone replacement results in agentic biology pipelines: swapping closed-source backbones with a BFT-aligned 70B model improves GeneAgent biological process reasoning and matches/exceeds the original VCWorld Gemini-2.5-Flash backbone on chemical perturbation reasoning (VCWorld average accuracy reported at 0.70 for BFT 70B vs 0.68 for Gemini-2.5-Flash; SFT/DFT replacements lag behind).
8. Sparse-reward RL compatibility is a key takeaway: after subsequent GRPO on Tahoe-100M with sparse binary rewards, SFT and DFT degrade, but all BFT variants improve (e.g., BFT 70B from 0.70 to 0.74 average on held-out VCWorld cell lines). The paper links this to richer mechanistic traces (more entities, more causal connectives, longer responses), which increases “credit assignment surface area” under sparse rewards.
9. Beyond generation, BFT aims to narrow the generative–discriminative split in computational biology: BFT-generated biomedical profile texts (encoded with a text embedding model) yield stronger gene- and cell-level representations, improving gene property prediction and gene interaction tasks, cell clustering, multimodal integration (scIB), and perturbation response prediction—sometimes rivaling or outperforming specialized biology foundation models in reported settings.
10. Practical considerations: BFT introduces only one main hyperparameter (window size g, default 256) and is reported robust across a broad range; it also shows reduced hidden preference transfer in a synthetic-data “subliminal learning” style safety test compared to SFT, staying closer to the base model’s behavior.
💻Code: github.com/TencentAILabHealt…
📜Paper: arxiv.org/abs/2511.21075
#LLM #BioNLP #ComputationalBiology #BiomedicalAI #FineTuning #ReinforcementLearning #SingleCell #PerturbationBiology #RepresentationLearning #AIAlignment

1
14
1,488

Apr 30
A new Special Issue opens for submission!
Title: Application of Symmetry in #NaturalLanguageProcessing
Editor: Sunday Olusegun Ojo and Olawande Daramola
Details: brnw.ch/21x23U2
#callforpapers #mdpisymmetry #representationlearning #largelanguagemodels
@DUT_Tweets
@UPTuks

3
2
46

🇧🇷 #LCS2 goes to #Rio 🇧🇷 Presenting our paper where we move beyond memoryless personalization → modeling user preferences as action-conditioned geometric walks with memory for better, user-aligned summaries. See you at #Riocentro 🚀 #Personalization #RepresentationLearning

Happy to announce that our paper has been accepted to #ICLR2026! 🎉
📜 Beyond Markovian Drifts: Action-Biased Geometric Walks with Memory for Personalized Summarization
👥 Parthiv Chatterjee, Asish Batha, Tashvi Patel, @sourish_rygbee, @Tanmoy_Chak
Congratulations to all authors!

1
3
462