
- Tweets 67
- Following 454
- Followers 1,175
- Likes 217






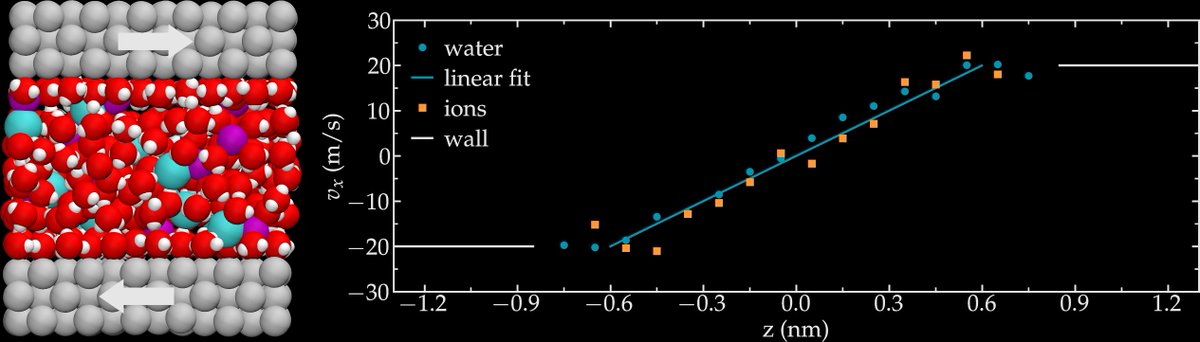

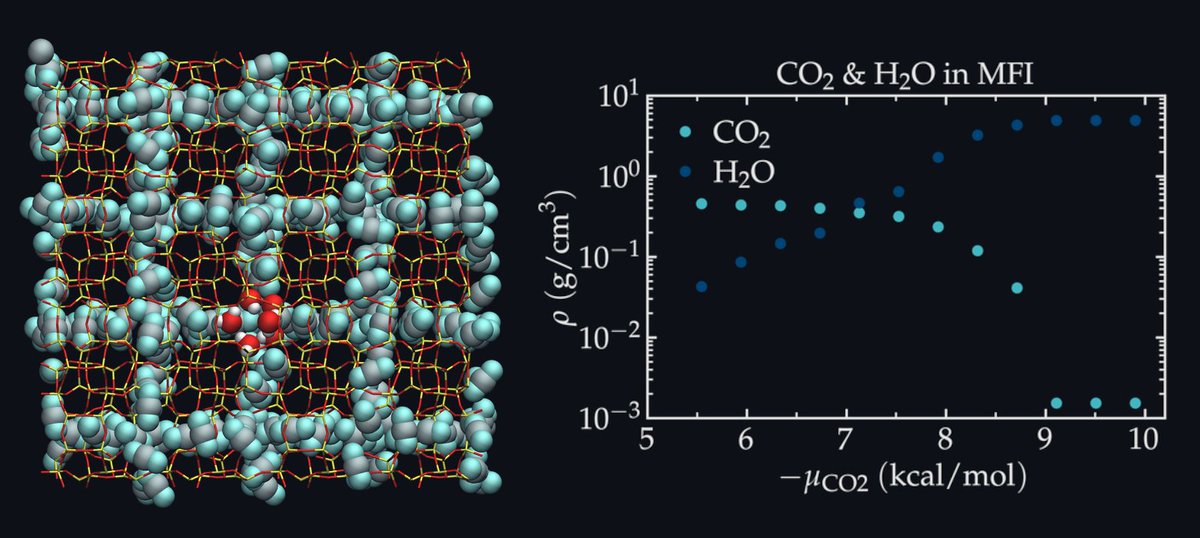
ALT LAMMPS snapshots of systems simulated during the tutorials.

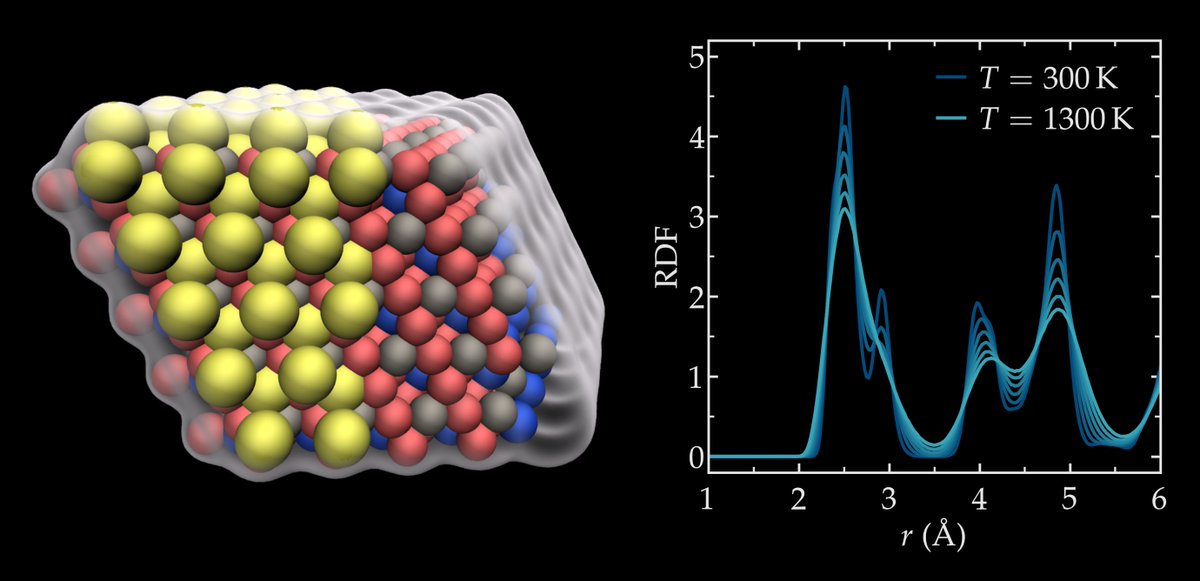
ALT In silico design and prediction of metastable quaternary phases in Cu-Ni-Si-Cr alloys
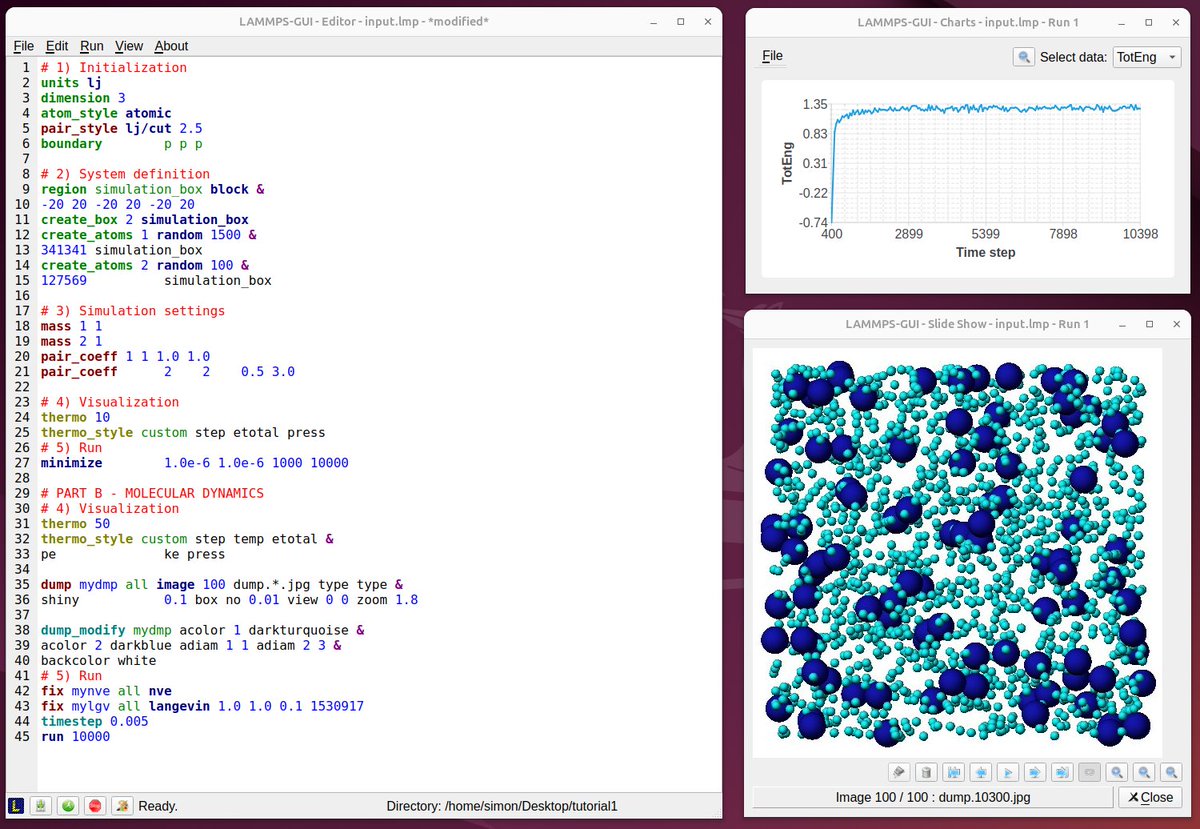
ALT Screenshots from LAMMPS tutorials, 3 years ago and today
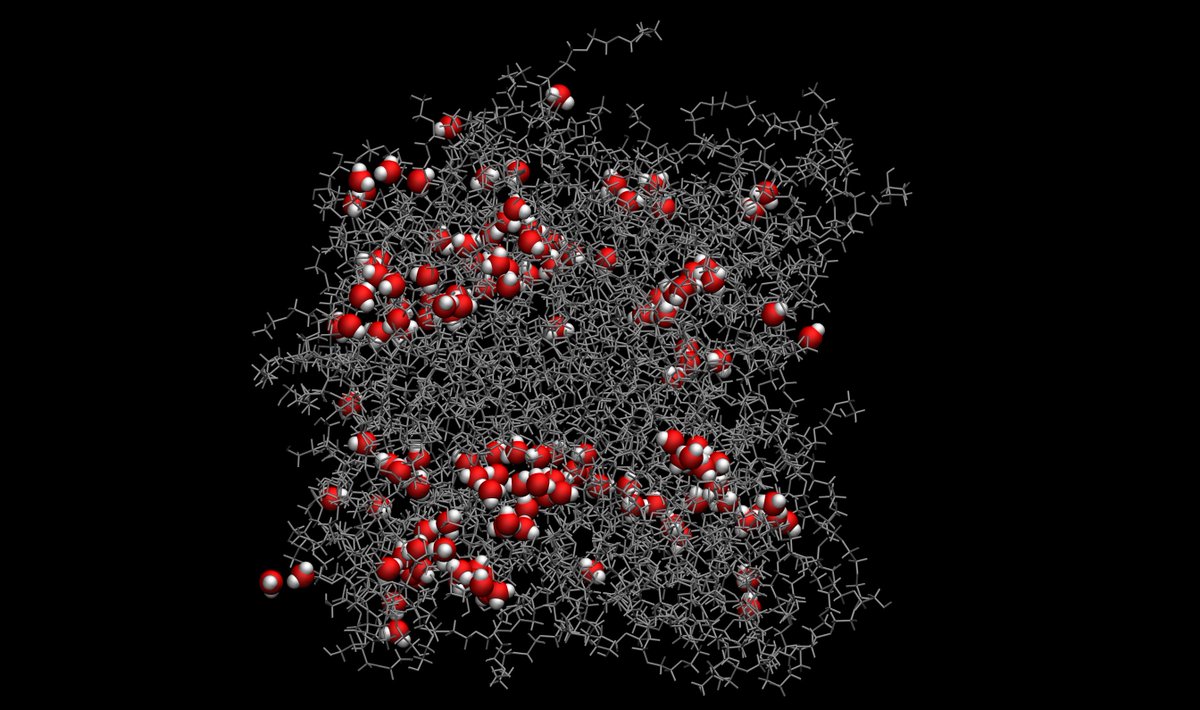
ALT Water molecules in a polymer
ALT Lennard-Jones fluid made of neutral particles with two different diameters : https://lammpstutorials.github.io/sphinx/build/html/tutorials/level1/lennard-jones-fluid.html
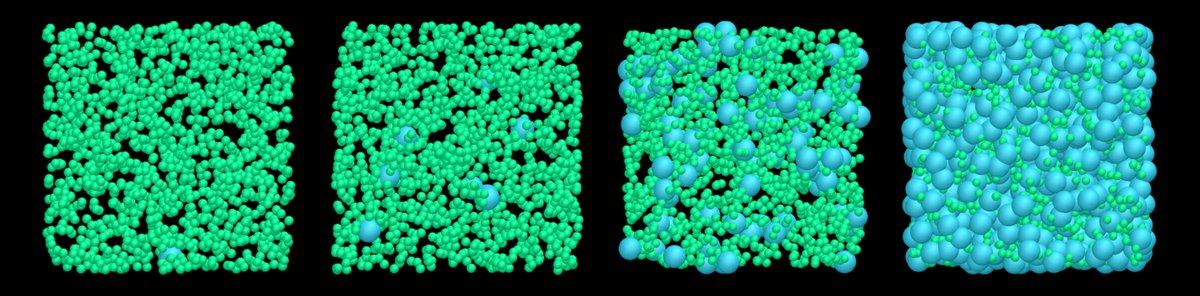
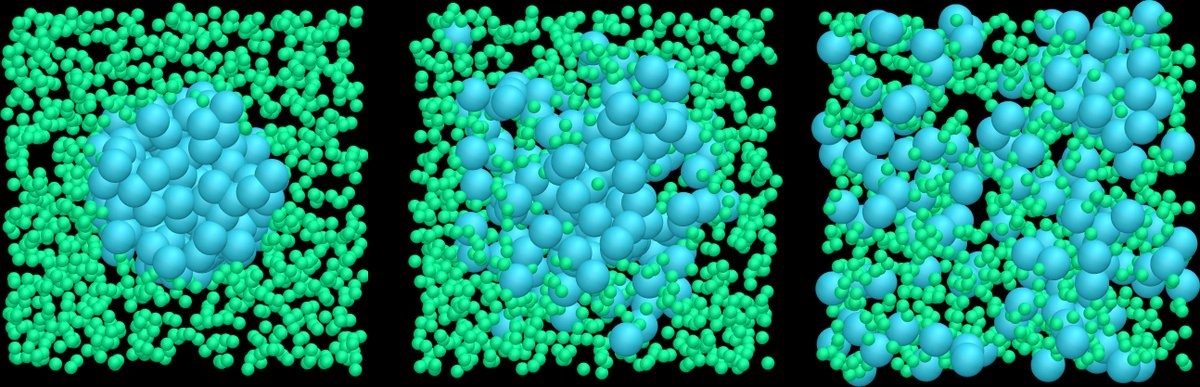
ALT Binary Lennard-Jones fluid with 1500 particles of type 1, and an increasing number of particles of type 2, from 1 (left) to 729 (right).
ALT Si (red), O (blue) and H (green) atoms.
ALT A simulated system rendered using Visual Molecular Dynamics (VMD)
ALT Left: electrolyte confined in a nanopore. Right: Velocity profile of both water and salt under shearing.
ALT Generated using ascii-image-converter ./logo.png -b --threshold 35 -W 80
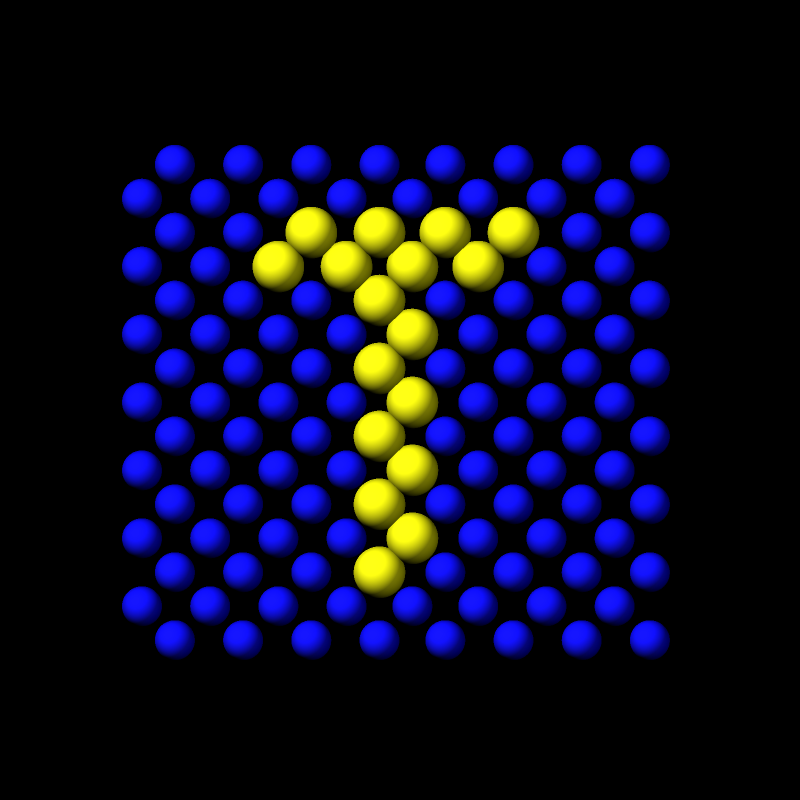
ALT Particles of different types mixing