
58 Photos and videos













What a story for all patients with pancreatic cancer
Always nice to see progress in biotech world
May 31

a standing ovation for daraxonrasib at asco. over 40k oncologists, entrepreneurs, investors, and patient advocates together celebrating revmed's breakthru in the fight against pancreatic cancer. u never forget these moments. it's what innovation is all about.
187
$celc
This is what intrigues me about celcuity
We have data in a few days with potential of 14 -15 months mpfs making this drug one of the best out there
More importantly, drug approval getting close so I am not sure how we are trading at $130 with imminent data and approval
May 1
3/ ASCO June2, PDUFA July17. The mutant data being positive means docs dont even need to check for the mutation, except for prognosis. I suspect docs will Rx mutant pts in July when drug approved, so this will jump start the launch.
2
9
2,698

Very good analysis Jay, I think breat cancer potential plus prostate and possibly lung etc can push this over $20b into 2027

$CELC Ok, folks, here’s the long-form post. Even though it’s long form, I’ll try to be as concise as I can to avoid taking up your time. Some calculations are omitted cuz it’s too hard to fully type them out on X.
Disclaimer: All of this is my personal opinion, not financial advice to anyone. Please do your own due diligence before buying the stock. I am not a biotech specialist or expert, have no MD or PHD behind my name (I have a JD and a BA in Statistics tho(not sure if that matters lol)). As you can see from my post history, I invest in a wide range of industries (oil&gas, banking, insurance, tech, semis, biotech, etc). My approach is to try to find mispriced companies with imminent catalysts that can drive a rerating.
Moving on to my thesis on Celcuity and more specifically, why I think mPFS for the triplet arm is likely to come in near or greater than 14 months. We all know that they released positive topline PR three weeks ago. Both the triplet (Geda Palbo Fulv) and the doublet(Geda Fulv) achieved statistically significant improvement in PFS compared to control(Alpel Fulv). We don’t know the actual numbers, but Stifel pointed out that current investor expectations for HR is <0.60. Ok, with this info in mind, I will start making my points:
1. The doublet stat sig hit implies at least a mPFS of 11 months for the doublet arm
We know the alpel Fulv had an mPFS of 7.3 months in BYLieve and 7.4 months in EPIK on post CDK4/6 populations. It is safe to assume that this arm should be performing in line with those two trials. Let’s say it does 7.4 in Vik1.
The doublet arm has an n of 50 and was designed to be exploratory and not part of the primary analysis. In statistical power calculations, sample size and treatment effect size are inversely related. We can roughly reverse engineer the statistical threshold of the log-rank test using the sample sizes (n = 50 for the doublet and n = 150 for the control) and a baseline control of 7.5 months. This allows us to calculate the minimum mPFS the doublet arm must have achieved to hit statistical significance(80% power or P<0.05).
Assuming 70% event maturity rate, the maximum threshold HR to hit p<0.05 is roughly 0.68. Assuming mPFS of 7.4 for control, the minimum doublet mPFS required to hit stat sig is approximately 11 months.
2. The delta between the triplet and doublet mPFS is likely to be at least similar to and maybe even larger than the 1.9 months delta observed in the WT trial.
So now we know the doublet arm has at least a mPFS of 11 months, what can that tell us about the mPFS for the triplet arm? Well, we know from the WT trial that there was a delta of 1.9 months between the triplet and doublet mPFS. The delta is entirely attributable to CDK 4/6 inhibition and proved that at least in WT tumors, gedatolisib's pan-PI3K/AKT/mTOR blockade effectively upregulates cyclin D-CDK4/6 signaling as a compensatory feedback mechanism, and adding palbociclib blocks this escape route, even after the patient has already progressed on a prior CDK4/6 inhibitor.
Well, this rebound in CDK4/6i activity portends even more favorably in the mutant population due to the simple fact that oncogenic PIK3CA mutations activate the PI3K pathway, which Geda inhibits. Since there was no PIK3CA mutation driving PI3K/AKT/mTOR pathway dependency in the WT trial, PAM pathway inhibition via gedatolisib should be stronger in the mutant cohort than in WT, given more pathway dependency. Stronger PAM inhibition should lead to stronger compensatory CDK4/6 upregulation feedback. Therefore, palbociclib should have more rebound activity to suppress in mutant vs WT. Based on this theory, the delta between triplet and doublet can potentially be greater than the 1.9 months observed in the WT trial.
To be conservative, let’s assume the doublet comes in at the minimum mPFS threshold of 11 months, and the delta between triplet and doublet comes in at 2 months. This gives us a mPFS of 13 months for the triplet. IMO, 13 months is likely close to the minimum mPFS that the triplet arm will likely show. A triplet mPFS of 13 months compared to 7.5 months mPFS for control will give a HR below 0.6, which beats current investor expectations according to Lifesci.
3. Ph1b triplet data revealed at ESMO last year look very encouraging.
At ESMO last year, Celcuity presented phase 1b data for the geda triplet. Notably, the WT phase 3 triplet mPFS came in very similar to the phase 1b data (9.3 months compared to 9.0 months in phase 1b on a n of 60). Therefore, I think the phase 1b data carry some predictive power about phase 3 outcomes.
In the PIK3CA-mutated subgroup, the triplet arm in phase 1b yielded median PFS of 14.6 months on a n of 30. With an intermittent dosing regimen, which is what is being used in Viktoria-1, the mPFS was even higher at 19.7 months on a n of 11. I don’t expect the final mPFS to come anywhere close to the 19.7 figure, but it is an encouraging data point with favorable implications for the upcoming phase 3 mutant data.
4. Trial results delayed by several months suggest slower-than-expected event progression in the experimental arms, which is a good sign, potentially suggesting longer mPFS.
The mutant cohort trial data were supposed to come before year-end 2025, but got delayed due to slower-than-expected accumulation of progression events. Celcuity originally expected enough events by Q4 2025, but didn't get them until this quarter. That's about a 4-5 month delay driven by patients simply not progressing as quickly as modeled. Since we know from both BYLieve and EPIK that Alpelisib mPFS is roughly 7.4 months, it is hard to imagine the control arm being the one outperforming and causing the delay. Therefore, it is most likely the experimental arms caused the delay, which means patients in the triplet/doublet arms are likely staying progression-free longer than anticipated. IMO, this is a strong piece of circumstantial evidence for triplet outperformance.
5. Late acceptance into the ASCO late-breaking line-up
Celcuity toplined the win in the PIK3CA mutant cohort of VIKTORIA-1 after hours on Friday, May 1. Celcuity was not in the April 21 late-breaking lineup when LBA titles were publicly released.
ASCO explicitly allows Phase III clinical trials for which final data are not available by the March 9 deadline to be granted an exception to submit later. However, the initial trial information must have been submitted by the January 27 deadline. ASCO does note that a later submission may have a negative impact on placement in the program.
My guess is that Celcuity used the Phase III exception. They submitted initial trial information by January 27, missed the March 9 deadline, then submitted data once complete, and ASCO granted them a slot. Overall, I think getting an ASCO oral LBA slot this late portends very positively for the data.
The following three points are akin to reading tea leaves, so take them with a grain of salt. Nevertheless, I still think they are worth mentioning.
6. Q1 conference call language compared to Q4 about the 2L market opportunity and what that possibly implies about mutant mPFS.
In both Q4 and Q1 conference call prepared remarks, Brian mentioned a greater than $5 billion market opportunity in the US for 2L HR /HER2- ABC and estimated $2.5 billion peak revenue using reasonable assumptions. The Duration of Therapy assumption management is using for 2L is around 12 months, based on the May investor presentation slide 11.
We know in WT triplet, mPFS is 9.3 months. We also know the mutant population accounts for roughly 40% of the total 2L population. Assuming equal penetration into both WT and mutant populations, we can try to calculate the mPFS assumption for the mutant population in order to reach an average Duration of Therapy of 12 months for the total population.
0.6*9.3 0.4*x = 12, which simplifies into x = 16.05, meaning the triplet mPFS assumption for the mutant population is roughly 16 months. Obviously, this is likely too simplistic, but I still think it gives a good ballpark figure.
What I think is interesting here is that they didn’t change/remove anything in the prepared comments about the market opportunity/peak sales potential between the Q4 and the Q1 call. Since they already have the mutant data in hand at the time of Q1 call, this gives me a small confidence boost that the mutant data is in line with their DoT assumptions, which were based on the Phase 1b data. I put much less weight on this point given its speculative nature.
7. Expansion of the Viktoria-2 1L trial to include ET sensitive patients
This is also an encouraging sign that potentially suggests that the mutant data is strong enough to justify including ET sensitive pts in Vik-2. The ETS arm is going to be a very lengthy and costly trial, given the DoT assumption. So I think management has to have pretty strong confidence in the PoS to be making such a move. They are going for the whole HR /HER2- Breast Cancer pie, which in total can generate over 10 billion in peak annual revenue for Celcuity. SubQ is also a huge deal, especially regarding IP duration expansion. Overall, definitely a positive sign.
8. Language used in positive mutant topline PR
“a transformative new medicine”, “significantly improve outcomes”, “improve the standard of care.”
Again, another reading of tea leaves kind of thing here. I don’t put much weight on this, but I do like the language being used here.
9. Safety
I am taking management at their word in the PR that there are no new safety signals, and Geda was generally well tolerated. I think the safety profile is likely to come in line with the WT cohort. Still a risk here, but I think it’s small (perhaps less than 10%) based on the WT safety profile, so I am willing to take it.
10. My prediction on how the stock would react at different triplet mPFS:
I think 13 months and HR<0.6 is enough to beat current investor expectations, and the stock likely will pop to $160 on that.
14 or higher with HR near or lower than 0.5 should drive the stock meaningfully higher, likely towards the $180 to $200 range, with the street/investors significantly increasing both their peak sales estimates for 2L and their PoS estimates for 1L.
I still have a lot more thoughts on the current valuation of the stock, but I’ll save them for a future post. Fully diluted market cap is pushing 9 billion, which is quite a bit larger than the 6 billion-ish market cap showing up on screeners. However, I think it is still extraordinarily cheap if mutant data comes in as positive as I predict (>14 mPFS), especially considering the larger 1L market opportunity. One point that is not talked about often enough is Geda’s unique positioning as the only successful/not-too-toxic PAMi on the market. They literally own this pathway to themselves with no me-toos anywhere in sight. Talk about unique MoA, eh (abvx, nktr fans assemble!) If not bought out, I can see this company growing to a 50 billion dollar market cap over the next few years, given the immense potential Geda holds in many other indications.
Thank you for reading!
5
571
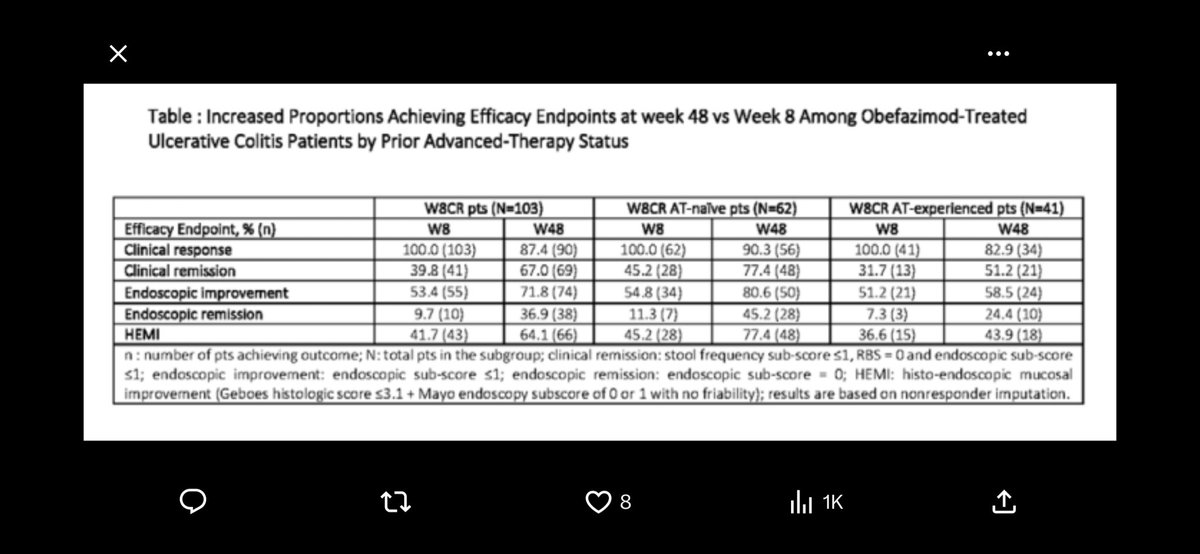
Great results by $CRVS
Looking forward to call at 130ET x.com/fortunoaurelar/status/…
3
309
One of my biggest issues with over trading
V well said
May 7

meh
honestly one of the things I've found over the years is that a lot of people look for quick results and that inevitably trickles down to their screen time, trades etc
Generally speaking, you have WAY more time than you think. Slow down and really think things through
2
4
384
A gift at 140

$celc adding more here low 140s hardly overbought, still more data at ASCO, pdufa in July or earlier. Anticipate this is a buyout by or before jpm 2027
3
591
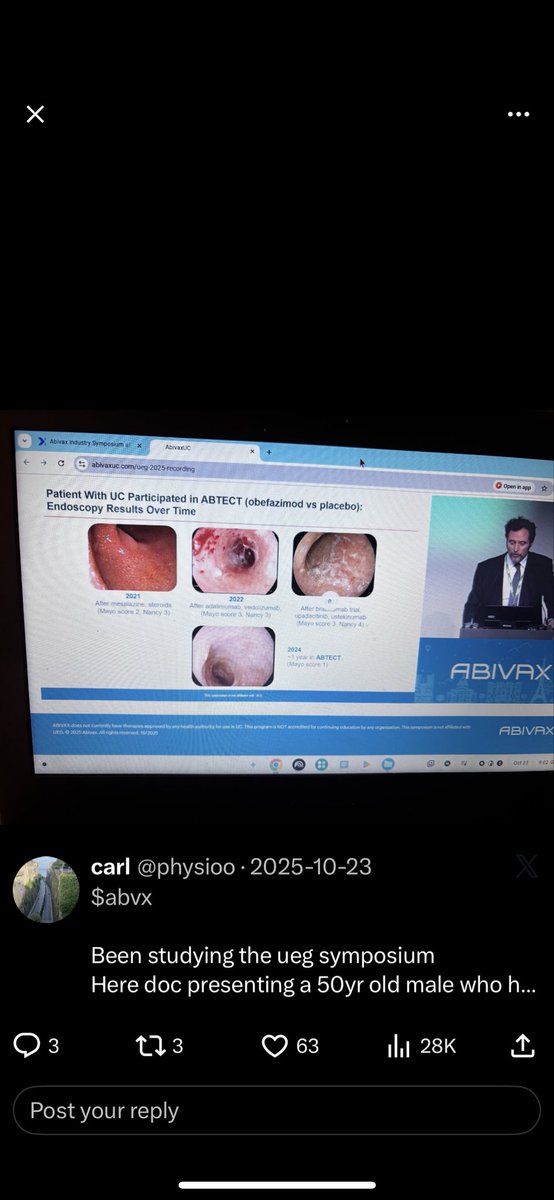
Impressive work here on $crvs
Looking forward
For upcoming data

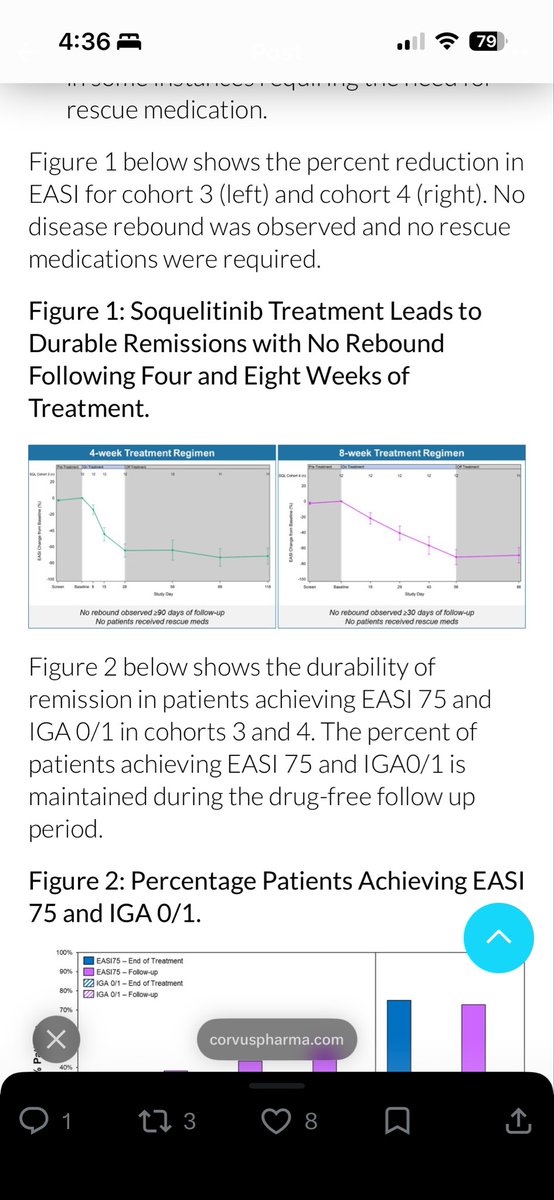
$CRVS Explanation I have been working on to explain #soquelitinib durability in atopic dermatitis w/ 90% of patients showing AD remission still 90 days post-treatment due to novel short-term Signal 1 interference v other Signal 3 blocking approaches = leading to "immune reset" and long-lived iTregs for durable disease control.
My hypothesis below fits w/ comments from Corvus they are seeing changes in JAK/STAT pathway and Tregs that they will disclose at SID in May.
My thoughts:
"A mechanistic hypothesis that selective ITK inhibition stabilizes induced FOXP3 Tregs through suppression of the IL-4/STAT6 epigenetic destabilization axis"
Background:
Induced regulatory T cells, or iTregs, are an attractive therapeutic concept in autoimmune and inflammatory disease because they offer the possibility of restoring immune balance rather than merely suppressing downstream inflammation. In principle, a durable increase in stable FOXP3 suppressive T cells could reset the relationship between effector and regulatory immunity and produce benefits that persist after treatment ends. In practice, however, iTregs are often unstable. Inflammatory cytokine environments can weaken FOXP3 expression, reduce suppressive function, and permit reversion toward effector phenotypes. This fragility is a central limitation of many immune-rebalancing strategies.
A particularly important destabilizing pathway is the IL-4/STAT6 axis. IL-4 promotes #Th2 polarization, but it also acts as an antagonist of iTreg differentiation and maintenance. Through IL-4Rα and downstream #JAK/#STAT6 signaling, inflammatory Th2 conditions can oppose the establishment of a stable FOXP3 program. This is relevant because durable Treg biology depends not only on transient induction of FOXP3, but on maintenance of an epigenetic state at the FOXP3 locus that supports continued transcription and resists inflammatory reprogramming.
Selective ITK inhibition raises a potentially important question in this context. ITK sits upstream in T-cell receptor signaling (Signal 1) and is especially important for programs that support inflammatory helper T-cell differentiation, including Th2 and Th17 responses. If selective ITK inhibition reduces Th2-driving circuitry and IL-4 production while also favoring Treg-skewed differentiation, it may influence both the generation and the long-term stability of induced FOXP3 Tregs. That possibility leads to my central hypothesis: selective ITK inhibition may stabilize induced FOXP3 Tregs through suppression of the IL-4/STAT6 epigenetic destabilization axis.
Mechanistic Model - The proposed model has two linked stages:
The first stage is differentiation bias. Selective ITK inhibition weakens proximal TCR signaling (i.e., CD28) and thereby reduces support for inflammatory effector programs, especially Th2 and Th17 differentiation. In parallel, this signaling shift may favor the emergence of FOXP3 induced Tregs. In this sense, ITK inhibition may function as a lineage-biasing intervention, moving the immune system away from inflammatory helper states and toward a more regulatory balance.
The second stage is stability. This stage may be the more important or most important for explaining durable benefit. The model proposes that selective ITK inhibition reduces GATA-3 (strongly shown by Corvus) and IL-4 output from effector T cells, which in turn lowers activation of the IL-4Rα-JAK-STAT6 pathway. Reduced STAT6 signaling then relieves a major source of iTreg destabilization. Under these conditions, induced FOXP3 Tregs may be more likely to preserve an epigenetic state compatible with durable lineage identity.
Next, three candidate regulatory nodes I have identified seem especially relevant in this framework. First, reduced STAT6 signaling may lower #DNMT1 activity or expression, helping preserve demethylation of the FOXP3 Treg-specific demethylated region and supporting continued FOXP3 transcription. Second, reduced IL-4/STAT6 signaling may decrease #HDAC9 activity or expression, favoring retention of histone acetylation across key FOXP3 regulatory elements such as CNS1 and CNS2. Third, histone acetyltransferase (#HAT1) activity may become relatively favored, potentially increasing chromatin accessibility at the FOXP3 locus. The combined effect would be a more transcriptionally open FOXP3 region, more persistent FOXP3 expression, and a more stable suppressive iTreg phenotype.
This model does not require ITK inhibition to bind directly to chromatin regulators. Instead selective ITK inhibition may act upstream of the epigenetic program by changing the cytokine and transcription-factor environment that determines whether induced Tregs remain stable or become plastic.
Supporting Evidence in Literature:
Several lines of evidence support this framework, although they do not yet establish the full mechanism.
1. ITK biology supports the differentiation component of the model. ITK is a key signaling node downstream of the TCR and has a recognized role in inflammatory helper T-cell differentiation. Studies of ITK-deficient or ITK-inhibited systems support the idea that dampening ITK can shift the balance away from inflammatory effector programs and toward regulatory phenotypes. Avery August paper even showed that CPI-818 (soquelitinib) inhibition of ITK act like a switch diverting Th17 destined effector T-cells to the regulatory iTreg phenotype. This provides a plausible basis for the idea that selective ITK inhibition may increase formation of induced FOXP3 Tregs in vivo.
2. The IL-4/STAT6 pathway is a credible destabilization axis for iTregs. IL-4 is not merely a Th2 cytokine; it also interferes with Treg differentiation and stability. Work on IL-4/STAT6 signaling has shown that this pathway can reduce Foxp3-associated chromatin accessibility and promote epigenetic conditions unfavorable for stable Treg identity. This makes STAT6 an especially meaningful intermediate between altered cytokine signaling and altered FOXP3 stability.
3. Epigenetic control of the FOXP3 locus is known to be central to Treg persistence. Stable Treg identity is associated with an accessible and appropriately demethylated FOXP3 locus, including key regulatory regions such as CNS1, CNS2, and the Treg-specific demethylated region (#TSDR). By contrast, transient or unstable FOXP3 induction is more vulnerable to loss under inflammatory conditions. This literature strongly supports the general principle that durable Treg function requires more than temporary transcriptional activation.
4. Prior work suggests that STAT6-deficient settings favor more suppressive Treg biology. Even Ibrutinib has shown to do this. Studies in STAT6-deficient systems have shown increased Treg abundance and the ability of relatively modest Treg increases to reduce allergic inflammation and impair recruitment of inflammatory effector T cells into tissues. Additional work has linked reduced STAT6 signaling to more favorable FOXP3-associated epigenetic states, including reduced DNMT1-related pressure. These findings do not prove the soquelitinib mechanism, but they strengthen the plausibility of the pathway being proposed.
5. Transcriptomic work in ITK-deficient T cells has identified changes in genes related to chromatin regulation, including HDAC9 and HAT1-associated signals. These findings are hypothesis-generating rather than definitive, but they support the idea that ITK biology may intersect with epigenetic regulators relevant to FOXP3 stability. In particular, the directionality of reduced HDAC9 and increased acetyltransferase-related signaling is consistent with a more open FOXP3 chromatin state.
Taken together, these strands support a coherent upstream-to-downstream logic: ITK inhibition may reduce inflammatory T-cell programming and IL-4 production; reduced IL-4 may dampen STAT6 activation; reduced STAT6 activity may lessen epigenetic pressure against stable FOXP3 expression; and the resulting iTregs may be more durable and suppressive than iTregs generated under inflammatory conditions.
What I am watching for that we may hear about at SID:
1. Whether selective ITK inhibition with soquelitinib reduces IL-4/STAT6 signaling in relevant immune compartments and pSTAT6 in the relevant human T-cell and Treg compartments to a biologically meaningful degree. This is central to the hypothesis.
2. Whether DNMT1, HDAC9, and HAT2 acetyltransferase-related pathways actually change in the predicted direction within induced Tregs generated under soquelitinib exposure.
3. Related to FOXP3 locus specificity including FOXP3 TSDR methylation analysis, chromatin accessibility testing such as ATAC-seq, and chromatin immunoprecipitation or related assays for histone acetylation across the FOXP3 promoter, CNS1, and CNS2, TSDR regions. This would establish whether the proposed upstream signaling changes actually produce the predicted local epigenetic consequences.
4. Whether any induced Tregs formed under ITK inhibition remain stable after drug withdrawal and inflammatory rechallenge. This is arguably the most important question of all. The hypothesis is ultimately about durability. It is not enough to show more FOXP3 cells on drug. It must be shown that these cells retain FOXP3 expression and suppressive function after washout, particularly when exposed again to destabilizing cytokines such as IL-4.
5. If data supports formation of stable iTreg lineage conversion, not merely transient FOXP3 induction. Many systems can increase FOXP3 expression temporarily without generating truly stable suppressive cells. A durable clinical effect would require stable iTregs, but Corvus already seeing a durable effect in atopic dermatitis which is highly suggestive they are.
The current framework is best presented as a mechanistic hypothesis with strong biologic coherence and literature support and high potential significance/benefit, but not yet as an established mechanism. Together, such data would convert the current model from a plausible hypothesis into a mechanistically supported explanation for post-treatment durability.
Conclusion:
Selective ITK inhibition may have effects that extend beyond transient anti-inflammatory suppression. A coherent mechanistic model is that it first biases T-cell differentiation toward induced FOXP3 regulatory states and away from inflammatory helper programs, then stabilizes those induced Tregs indirectly by suppressing the IL-4/STAT6 axis that normally drives epigenetic destabilization. In that setting, reduced DNMT1 and HDAC9 pressure, along with a more accessible FOXP3 locus, could help maintain persistent FOXP3 expression and durable suppressive function.
The significance of this hypothesis is HIGH because, if validated, it would offer a biologically plausible explanation for benefit that persists beyond active dosing. It would also position selective ITK inhibition as a true "immune-rebalancing strategy" rather than simply another anti-cytokine or downstream suppressive approach that reverses as soon as treatment is stopped.
If my hypothesis is validated, it is potentially game-changing not just for atopic dermatitis but for a broad range of I&I indications because it would move soquelitinib out of the standard “anti-inflammatory suppressor” bucket and into an “immune reset/stability-restoration” bucket. That is a much bigger claim. Scientifically, it would suggest that ITK inhibition is upstream enough (Signal 1) to alter not just cytokine output but the long-term lineage stability of suppressive T cells. Therapeutically, that would support intermittent dosing and off-drug durability rather than simple chronic suppression. This would be a revolutionary approach and paradigm shift in treatment of auto-immune diseases.

1
7
1,303


Nice work

$celc by the numbers
fdmc: $9.3B @ $140/sh. cash: $400M (offering coming)
valuation: base --> bull case - $15B ($225/sh) - $40B ($600/sh)
catalysts: 1.) ph3 full data (mpfs/hr) at asco - june 2, '26. need mpfs: 10m / hr: <0.60 / safety.
2.) 2l wt bca pdufa - july 17, '26.
3
739