
Microbial ecologist/bioinformatician PhD 👽 | Bioinf-beginners go to microbialomics.org | For phylogenomics, GToTree :)
Joined May 2015
- Tweets 4,904
- Following 3,226
- Followers 3,289
- Likes 6,521
437 Photos and videos











Pinned Tweet

29 Jun 2024
New music alert!!
In my pre-science life I used to make music 🎸🎤🎵 And I just put out a new song!
The link below will get you to your preferred streaming/music service. Check it out 🙂
onlyonce.hearnow.com
3
2
6
1,722
Mike Lee retweeted

Jun 10
Arguably the most boring step in genomics is the first one: normalization. Settled science. Scale log. Move on.
Except that here's been a huge blind spot in the field. And it matters for AIxBio. A 🧵about what I think may be one of the most important papers I've written. 1/
18
147
670
114,055
Mike Lee retweeted

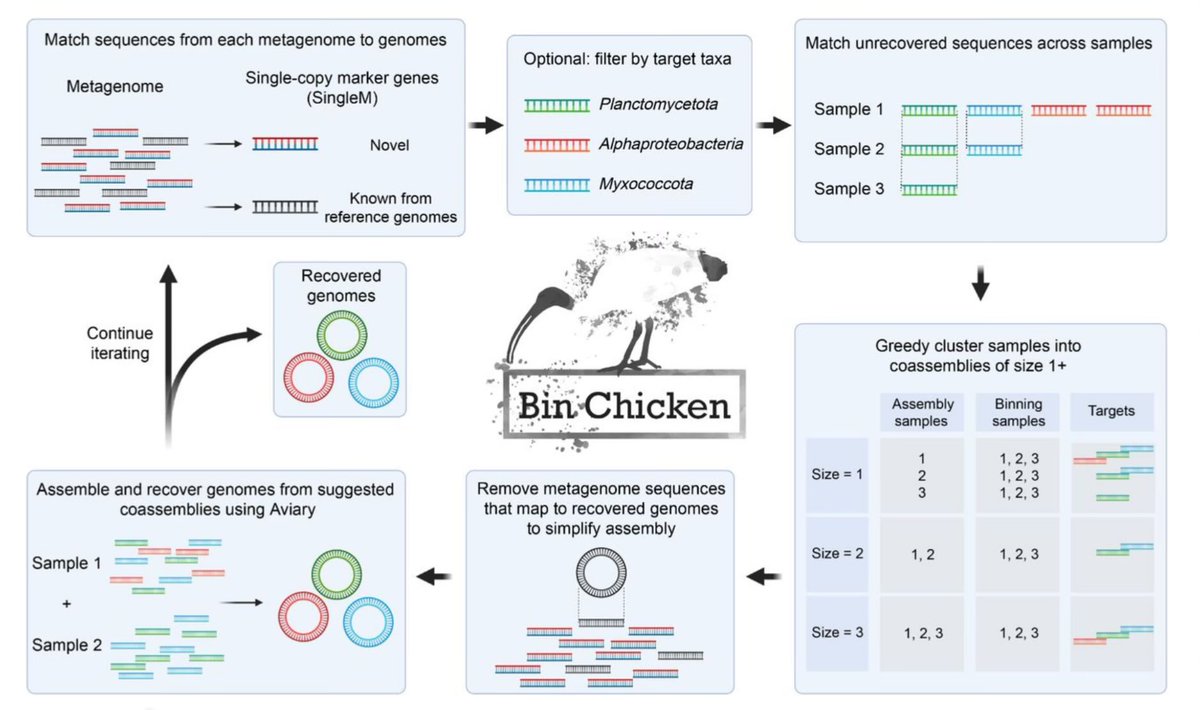
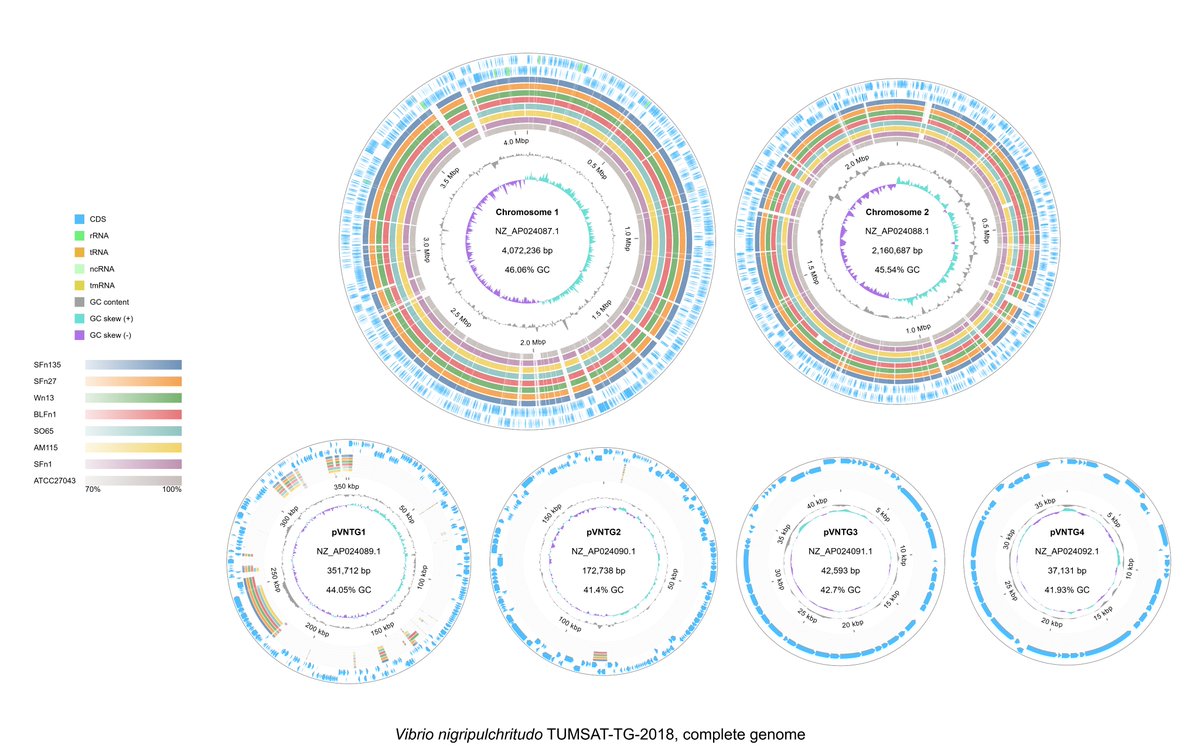
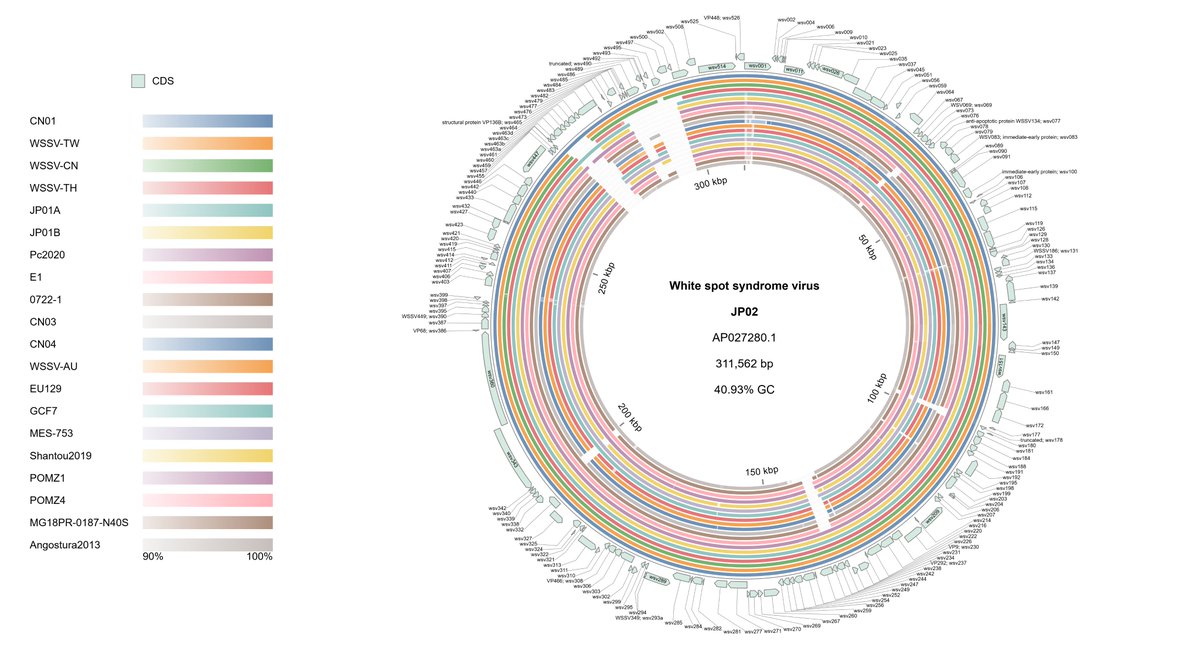
Circular genome comparison is now live in #gbdraw! 🚀 The built-in LOSATN instantly runs pairwise alignments and generates homology rings for your sequences, 100% in your browser! Try it out: gbdraw.app #bioinformatics #genomics #microbiology
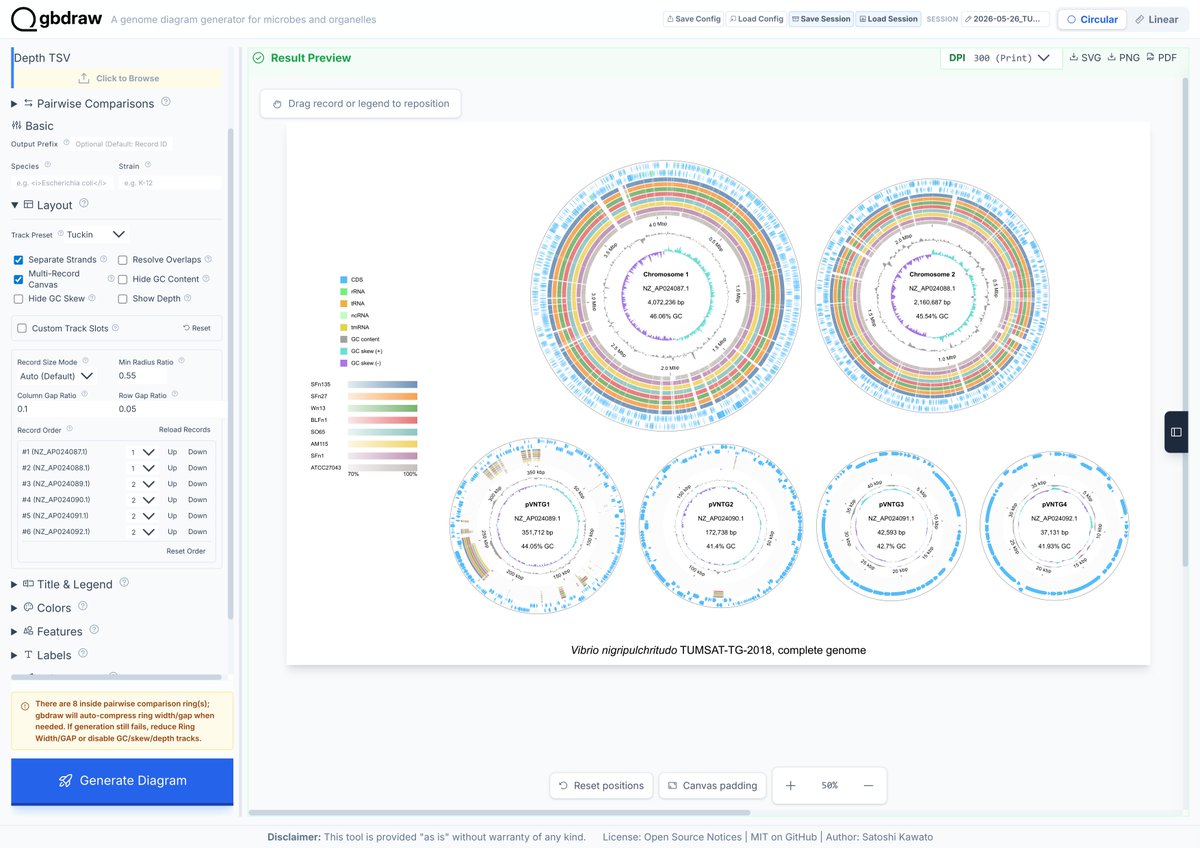
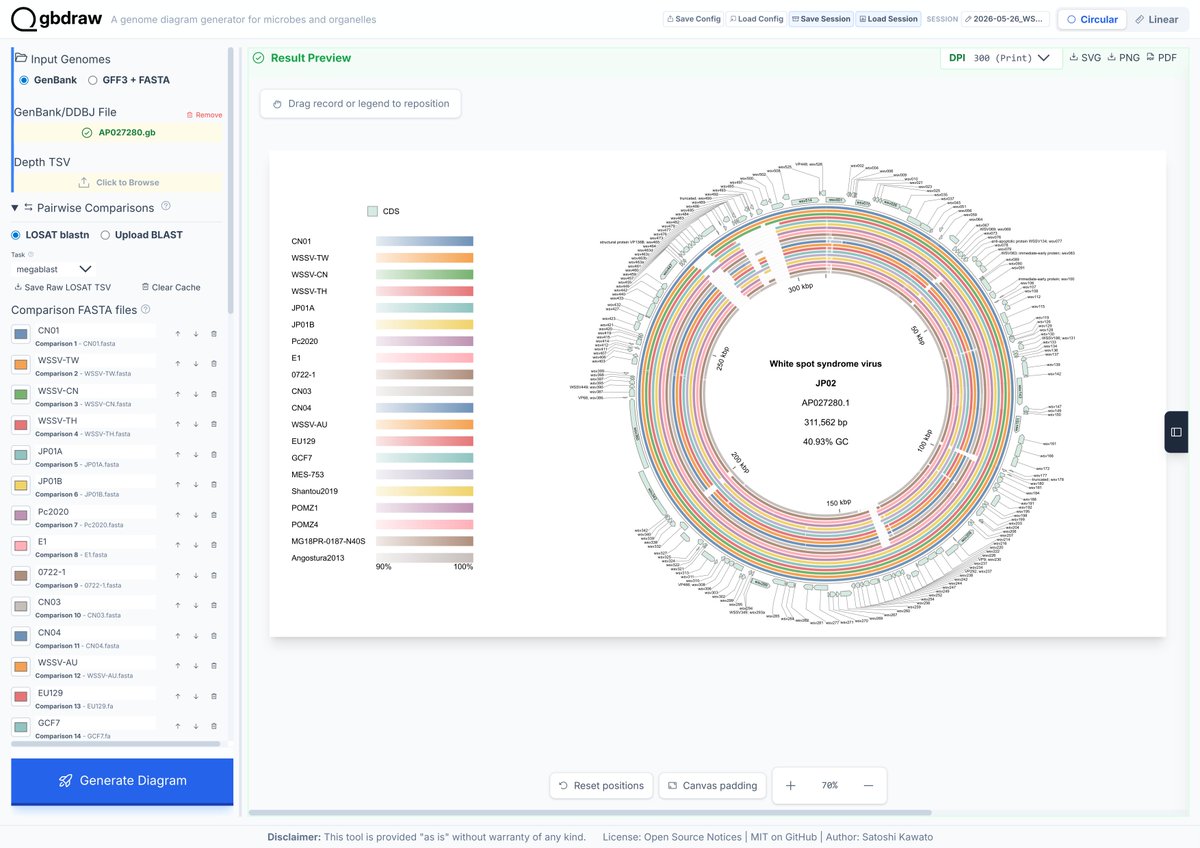
1
42
148
9,063
Mike Lee retweeted

May 20
How do you publish this tree and claim it's a success???!!!

May 19

We are releasing Carbon: a crazy fast DNA model
Carbon is 275x faster than the next best model. So fast you can process the whole human genome on a single GPU in <2 days.
Here are the tricks we used:
When modelling DNA sequences a lot of the performance comes down to tokenizing the sequences in a smart way. BPE tokenizer struggle because there are no whitespaces and character (called base in DNA) level tokenizers waste a lot of compute on too many tokens.
Carbon is built with a unique tokenizer: we split sequences in chunks of 6 bases, but during both training and inference we can work with single base resolution. That's similar to having word tokens but resolving them at the character level. All possible thanks to the DNA tokens unique structure.
The architecture combined with the tokenizer makes the model 275x faster than the previous SoTA (Evo2) at this size.
We built an interactive demo so you can explore how the model can generate DNA sequences, investigate the structure of genes, predict the effect of mutations, generate and fold proteins and even reconstruct parts of the tree of life.
huggingface.co/spaces/Huggin…
35
44
397
88,323
Mike Lee retweeted

May 21
Microbe Decoder uncovers functional traits of microbes in microbiome datasets academic.oup.com/nar/advance…
27
71
2,958
Mike Lee retweeted
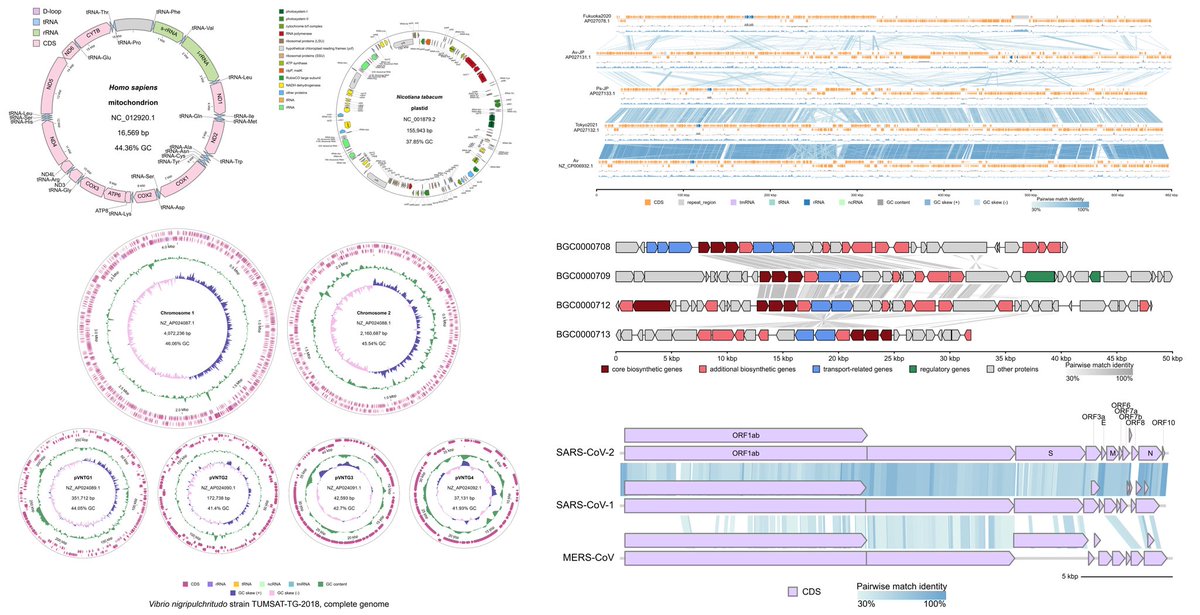
🧬Collinearity Analysis is here in #gbdraw v0.11.0! You can easily detect and visualize synteny and collinearity blocks using LOSATP. You can instantly align the entire diagram based on a shared ortholog!
Try it now: gbdraw.app/
#bioinformatics #genomics #microbiology

ALT Collinearity analysis of Hepatoplasmataceae, a group of mollicutes associated with isopod crustaceans. Inverted syntenic blocks are colored in dark red.
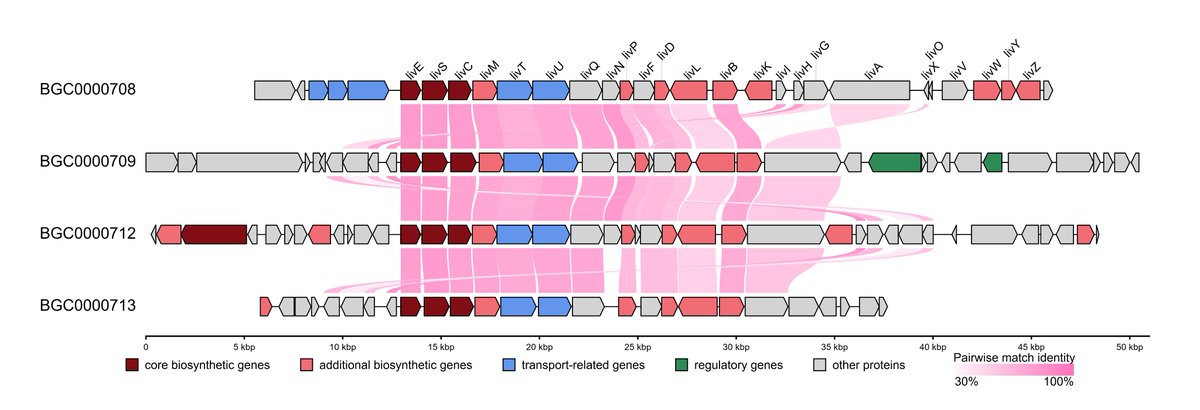
ALT Synteny of lividomycin-like aminoglycoside biosynthetic gene clusters. The diagrams are aligned based on livE gene (see next figure for the command)
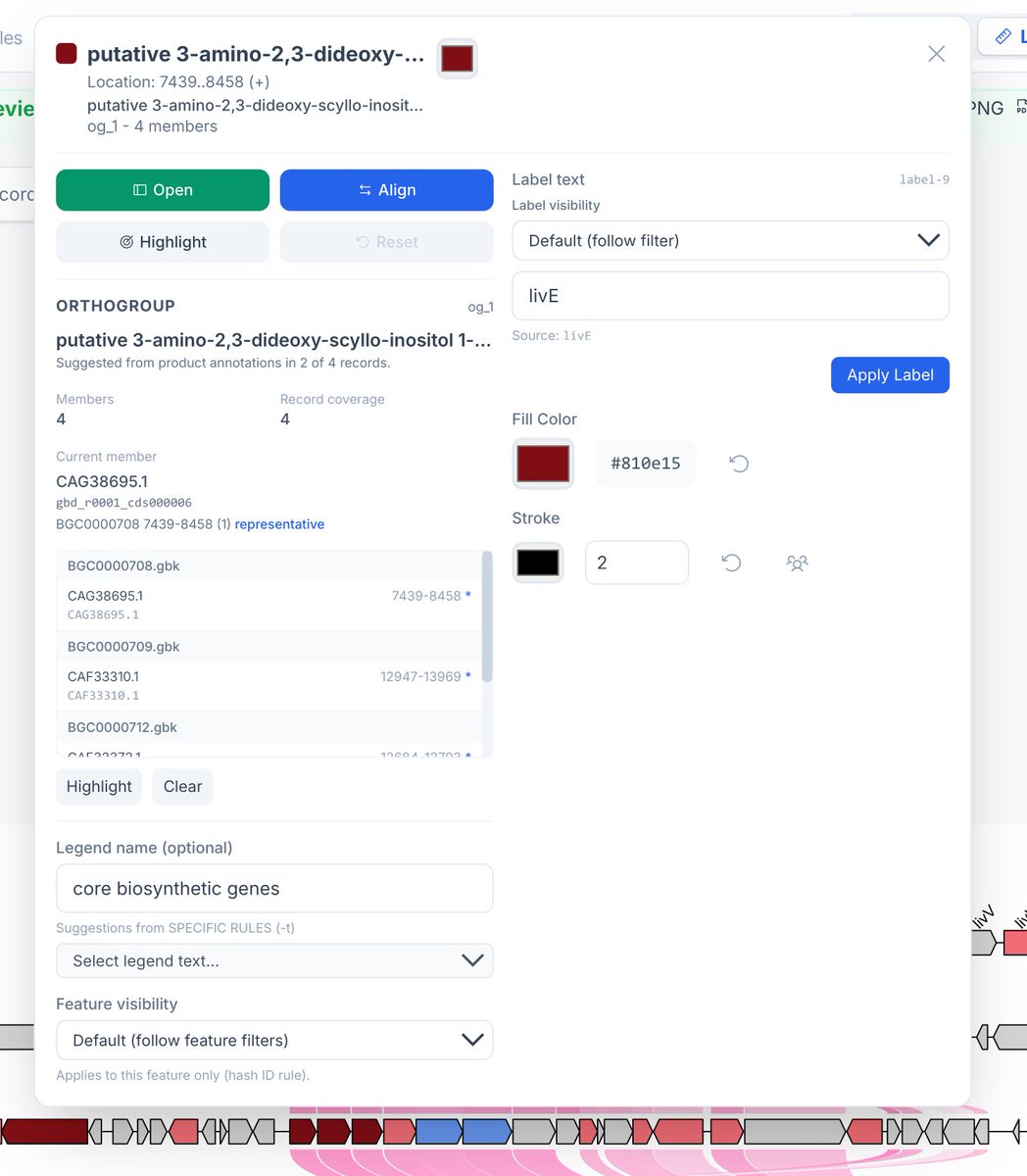
ALT Dialog box for the livE gene in the record BGC0000708. Click "Align" to align the records based on the shared livE orthologs, as seen in the previous figure.
1
49
171
9,717
Mike Lee retweeted

Welcome @aphillippy to @JohnsHopkins in both @JHUCompSci and @JHUBME, as a new @JHU_BDPs professor starting today! linkedin.com/posts/aphillipp…
1
4
27
3,699
Mike Lee retweeted

The DeepClust database, with 19B protein sequences clustered into 544M non-singleton clusters, hwas finally published after 3 fucking years. A lot of improvements will come from using these sequences in our analyses :)
paper
nature.com/articles/s41592-0…
DB
objectstore.hpccloud.mpcdf.m…

8 Sep 2025

I find this funny-sad. On July 25 GigaRef was introduced as the largest protein seq DB to date with 3.34B seqs. But on Sep 1, the LOGAN DB was released with >100B seqs. This means the title of the largest seq DB only lasted ~1month. Meanwhile the DeepClust DB was never released☠️
4
15
85
9,397
Mike Lee retweeted
Apr 16
For anyone coming upon this work for the first time (it was originally published two years ago on @biorxivpreprint before being republished by @nature this week), it is already the subject of extensive discourse (c.f. x.com/SashaGusevPosts/status…) and multiple follow-up papers (e.g. biorxiv.org/content/10.64898…) which are essential to understand when thinking about this paper.
Apr 16

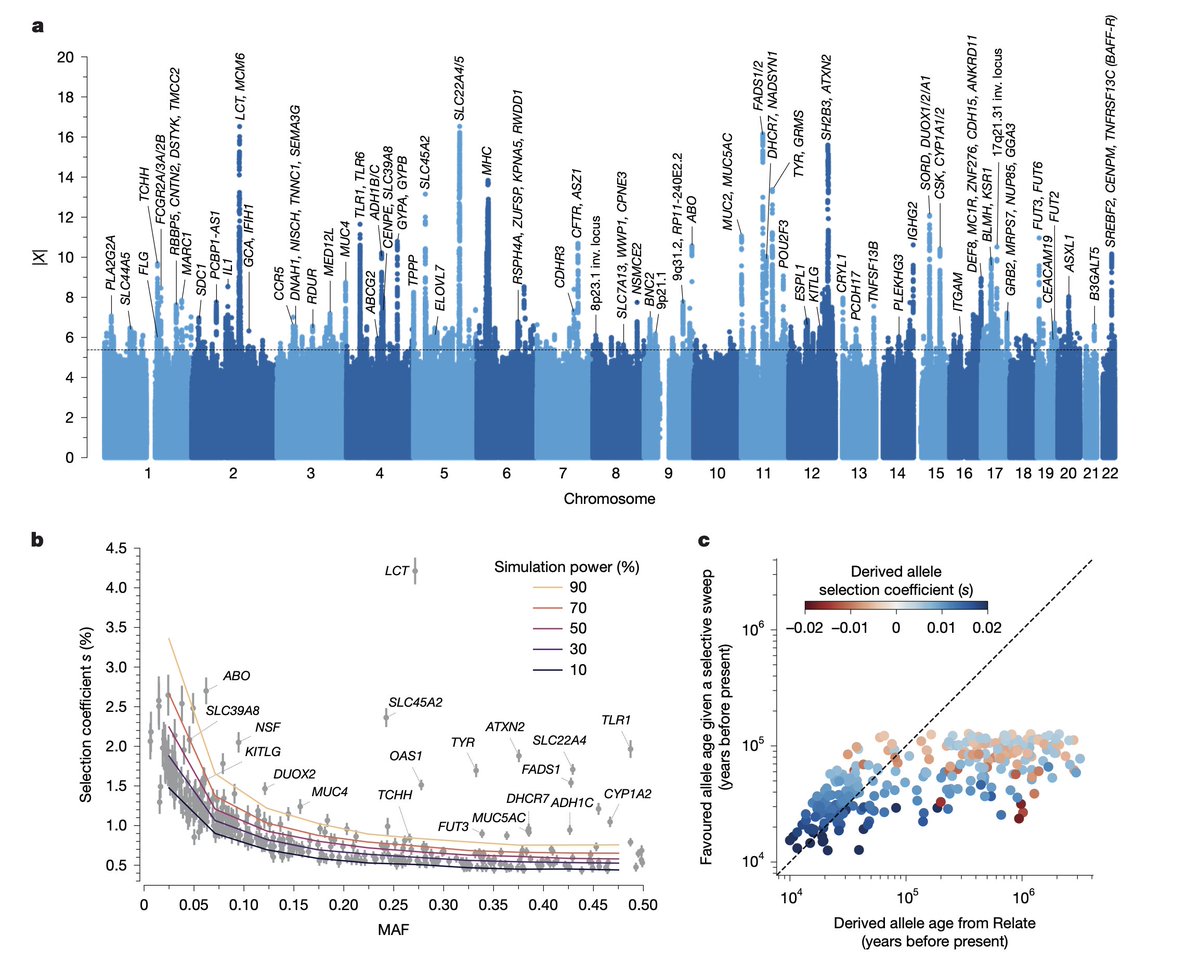
A new paper in @Nature from David Reich, @aliakbari23 and colleagues breaks the conventional understanding of recent human evolution. The field believed that strong selection in the recent past (~10,000 years) was rare, with few exceptions like the lactase persistence locus. In this paper, the authors challenge that belief, showing that we weren't looking at the problem right.
Previous studies that looked for evidence of selection using ancient DNA addressed the problem cross-sectionally, asking if allele frequencies differed across populations more than what one would expect based on genetic drift and migration. Most arrived at the conclusion that population structure primarily explained the observed differences. Here, the authors addressed the problem longitudinally, accounting for when ancient individuals lived by explicitly modeling time as a variable in the analysis. It turns out doing it this way dramatically increases power, increasing the number of genome-wide significant selection signals by 20-fold!
Looking at why accounting for the time variable led to such dramatic changes in results, the authors find that previous studies missed so much because selection often happened not on new variants leading to dramatic sweeps (the conventional model: new variant -> selection -> increase in frequency) but on already existing variants driven by transient environmental pressures. Many of these variants underwent reversals, selected up when a pressure existed, then purged when it disappeared or the trade-off cost became dominant. A great example is the TYK2 variant, where an allele boosting immunity was selected for thousands of years because it protected against TB, then got purged as TB endemicity declined and the autoimmune cost took over.
The scale of what they found is striking: hundreds of loci showing strong selection in the past 10,000 years with a median selection coefficient of ~0.86%. This number is pretty big in evolutionary terms, meaning allele frequencies have been shifting by ~1% per generation in a consistent direction. Previous selection scans found a maximum of 20 loci, and this one finds hundreds. That isn't an incremental change. It fundamentally reframes our understanding of how common strong selection has been in recent human history.
Some of the most striking findings come from polygenic selection, where hundreds of small-effect alleles were pushed in the same direction simultaneously. Polygenic scores based on large-scale GWAS of today predict recent negative selection for traits like body fat, waist circumference and schizophrenia, and positive selection for others like cognitive traits. One important caveat is that GWAS phenotypes are measured in industrialized societies today, and how well they capture what was actually being selected in ancient environments is debatable.
For me personally, these findings have direct implications for drug discovery. When using human genetics to find drug targets, we often fixate on the benefit and risk profiles of variants visible today. But we need to be aware that a variant's benefit:harm ratio might be environmentally contingent, and could reverse when the wrong environment manifests. An evolutionary understanding of a variant's association with traits is therefore essential.
The same logic applies, perhaps even more urgently, to embryo selection. Selecting embryos based on polygenic traits is humans making permanent, heritable decisions for their offspring with a narrow view of today's environment. The ancient DNA record now shows that cost-benefit landscapes flip over time. So, an embryo carrying man-made selections is carrying those changes into an unpredictable future environment.
The broader takeaway is that human evolution didn't freeze in the last 10,000 years. We just lacked the tools and datasets to see its movement. The current findings are based on European populations. I am curious to see these analyses extended to other populations too, like South Asian, East Asian and African populations, which might be holding more surprises to blow our minds.
Akbari et al. Nature 2026
nature.com/articles/s41586-0…
29
157
42,799
Mike Lee retweeted

How every layer of science's "self-correcting machinery" failed when Iva Veseli and I simply wanted to reproduce the findings of a high-profile study on gut microbiome and autism:
merenlab.org/2026/04/15/unfa…
2
22
109
19,662

Hungarian Prime Minister Viktor Orban conceded defeat to Peter Magyar in Sunday’s parliamentary election, according to the opposition leader bloomberg.com/news/articles/…
31
49
152
26,254
Mike Lee retweeted

Apr 8
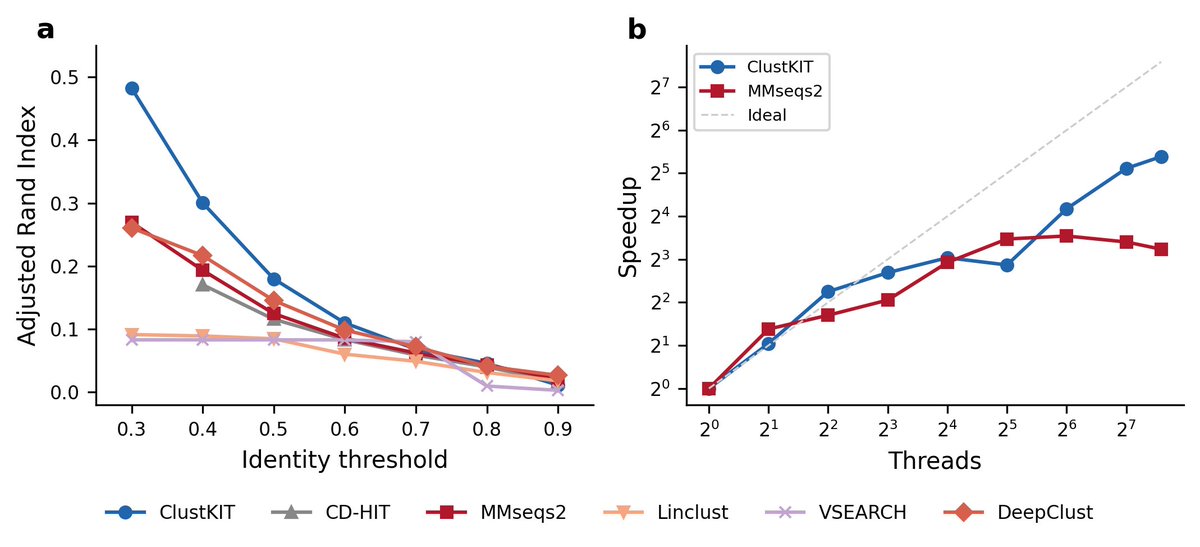
NEW tool: introducing ClustKIT, an accurate #protein sequence clustering
ClustKIT performs particularly well in the twilight zone of #sequence identity (L panel; low identity thresholds; Pfam benchmark w/ 22,343 sequences from 56 families)
ClustKIT also scales well (R panel)

3
14
31
4,602
Mike Lee retweeted
gbdraw v0.9.1 is out now on Bioconda!
Easily visualize microbial genomes and enjoy the #gbdraw web app completely offline.
📦 Install via Bioconda: bioconda.github.io/recipes/g…
🌐 Try the online app: gbdraw.app/
#bioinformatics #genomics #microbiology #visualization
2
88
300
15,966
Mike Lee retweeted
In my latest column, I explain some of my reasons for being deeply skeptical about AI models that claim to understand DNA, genes, and genomes: stevensalzberg.substack.com/…
6
32
155
16,501
Mike Lee retweeted

Mar 29
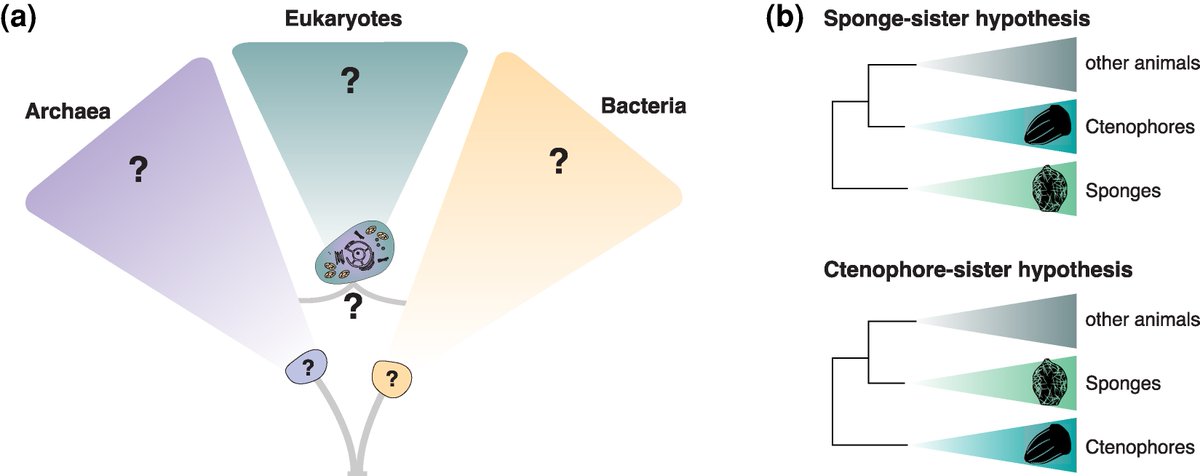
Toward a genomic understanding of the tree of life
#microbiology #evolution #eukaryotes #TreeofLife
doi.org/10.1093/molbev/msag0…
19
86
3,635
Mike Lee retweeted

Mar 29
This paper took us 13 years and is one of the longest papers ever in Cell. Check it out & judge for yourself
cell.com/cell/fulltext/S0092…


Just because epigenetic drift follows a predictable pattern doesn't necessarily mean it's causal in aging. Predictable patterns can form from stochastic reactions. Also you cannot fix all extracellular damages with cellular rejuvenation. More here: olafurpall.substack.com/p/wh…
25
270
1,512
224,653

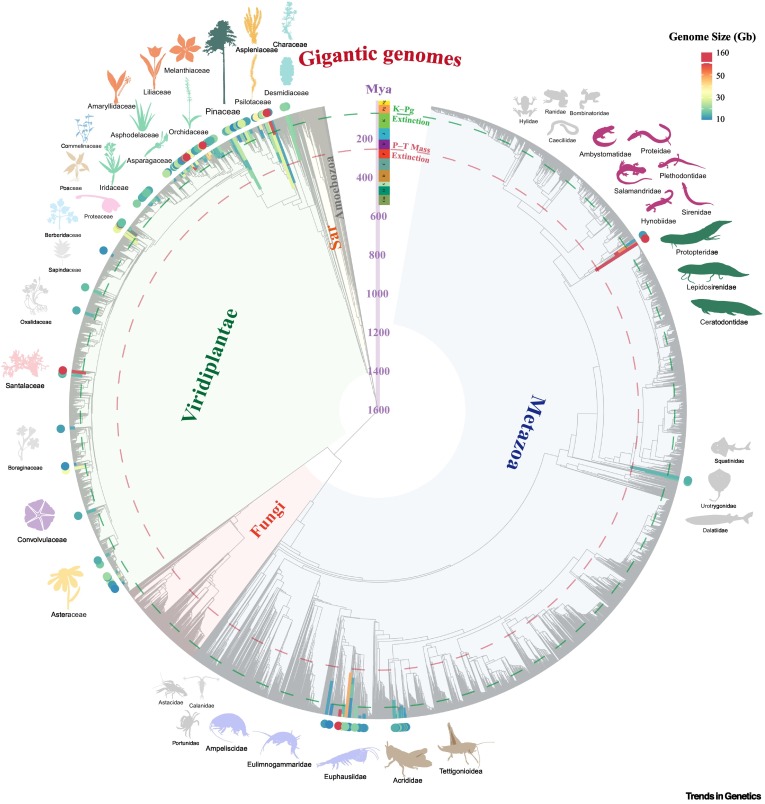
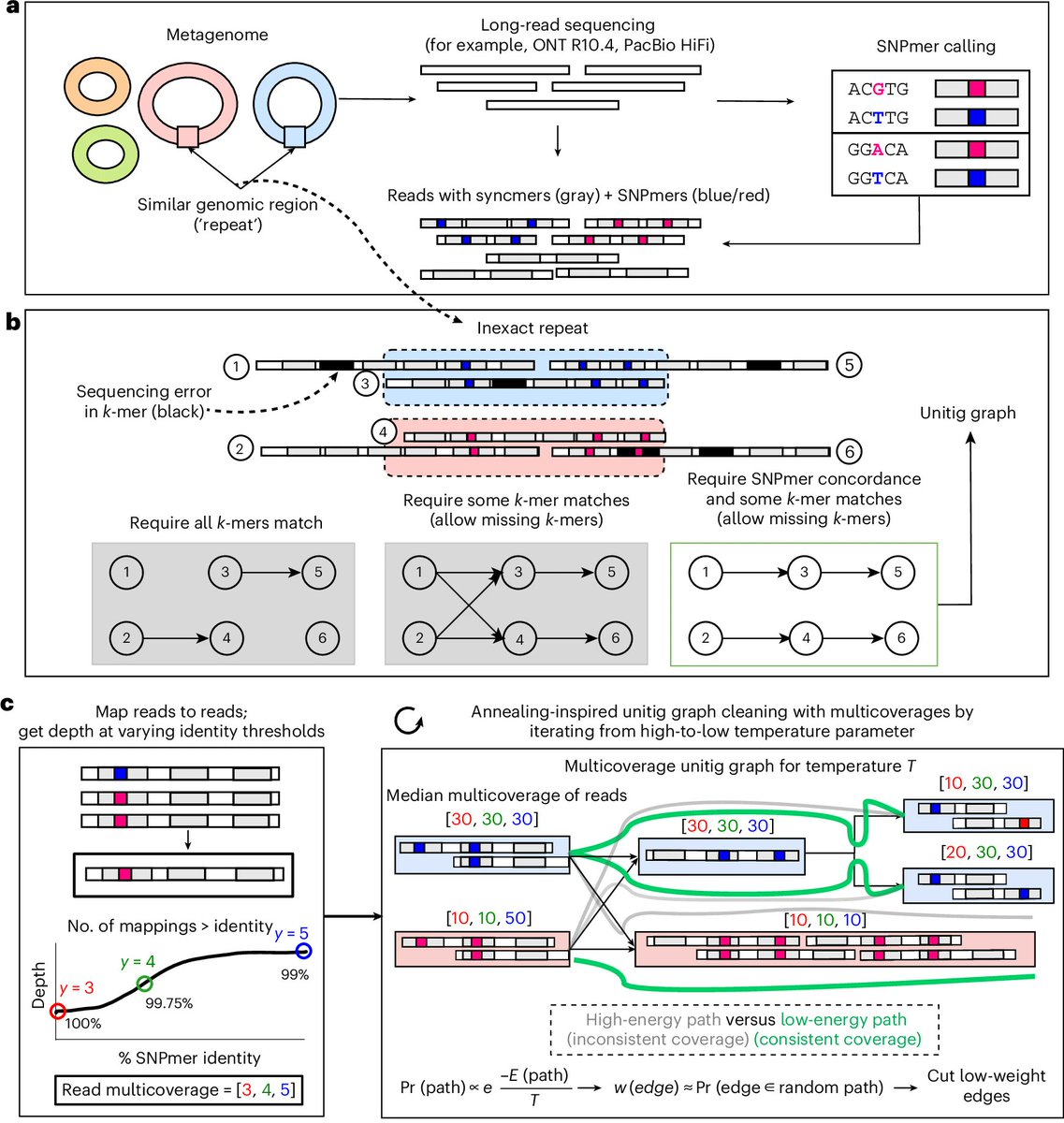
Mike Lee retweeted

Mar 27
Myloasm, our long-read metagenome assembler, is now published! w/ Max Marin & @lh3lh3
Very rewarding after > a year of development and countless hours thinking about assembly. Thanks to beta testers, Li lab, and reviewers for helpful feedback.
Link: rdcu.be/famFj

High-resolution metagenome assembly for modern long reads with myloasm - @lh3lh3 go.nature.com/3PBEwvR
1
36
95
21,556
Mike Lee retweeted
Mar 24
Clustering the protein universe of life using DIAMOND DeepClust nature.com/articles/s41592-0…
6
17
1,060
Mar 21
RT @PeterHotez: Seriously? I make low-cost vaccines that reached 100 million people, bypassed big pharma, didn’t make a dime, and saved 300…
2,388
Mike Lee retweeted

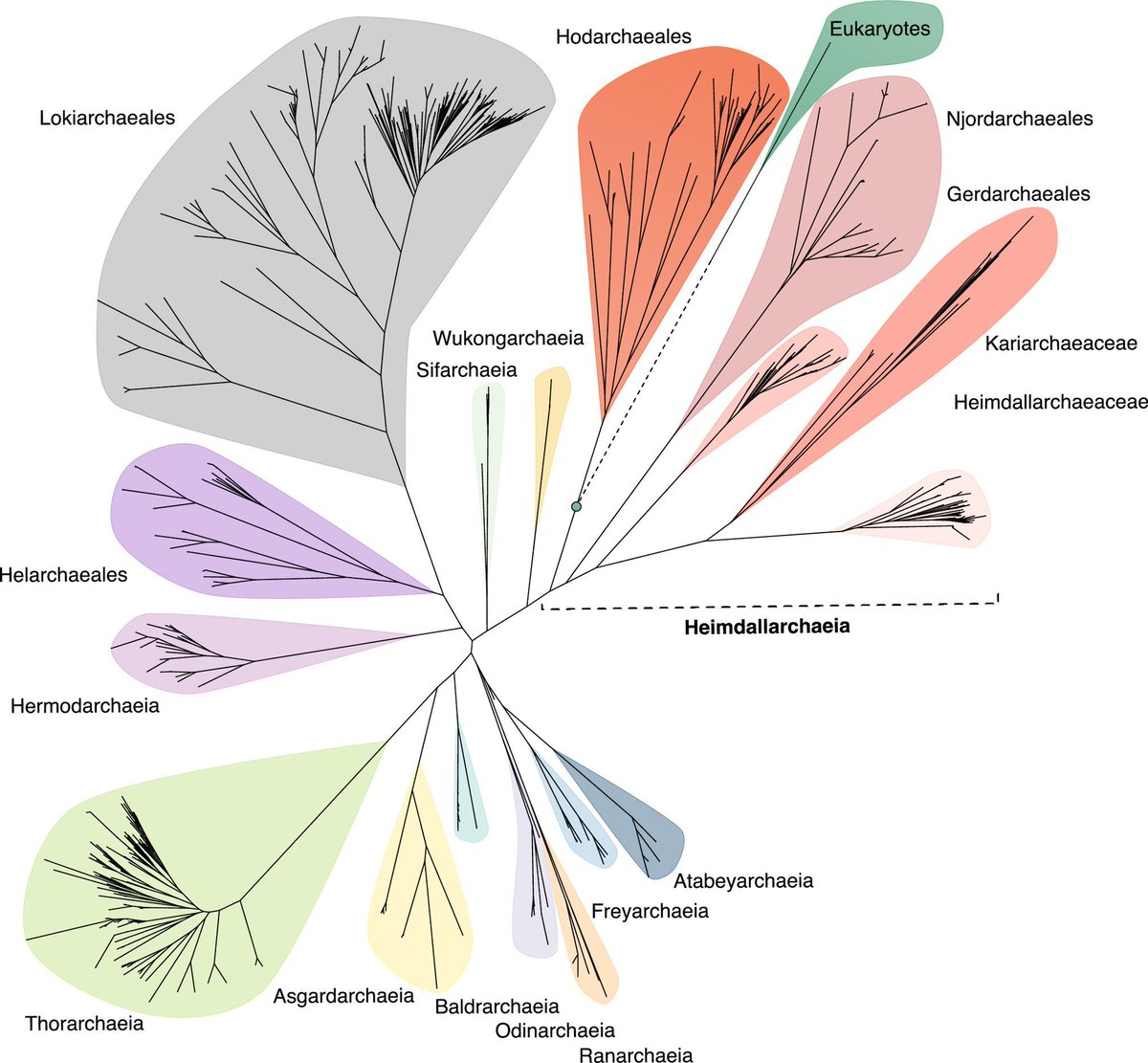
The archaeal roots of eukaryotic life pnas.org/doi/10.1073/pnas.25… 🧬💻🧪
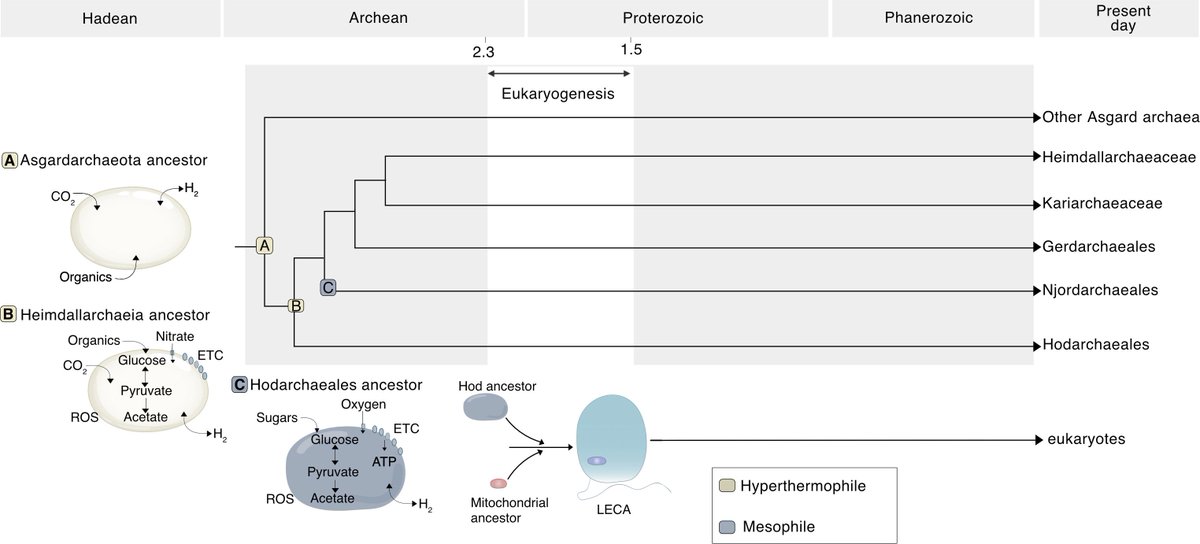
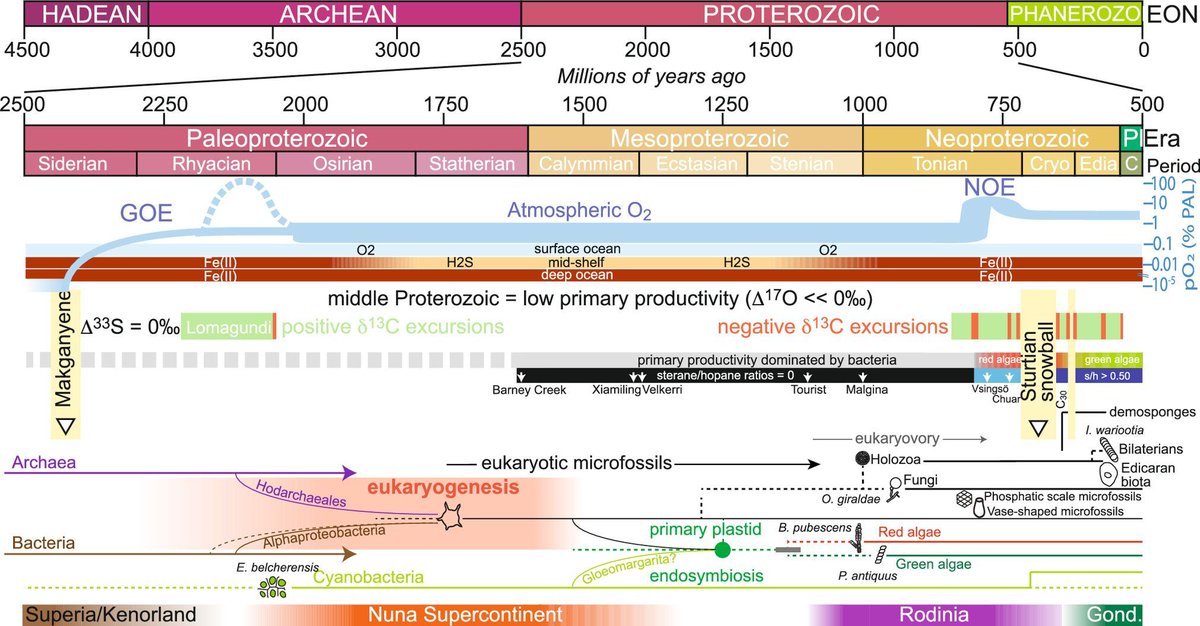
40
161
6,762