
Heme/onc fellow @PennCancer | @YaleMed MD/PhD w/ @sidichen | Harvard '16 | ryanchow.org
Joined October 2019
- Tweets 106
- Following 558
- Followers 352
- Likes 855
17 Photos and videos


Ryan Chow retweeted

Feb 3
Real-World Analysis Supports Rethinking Upfront Dose Intensity of Enfortumab Vedotin in Advanced Urothelial Cancer @ScienceChow @PennCancer @Ron_cology @ramsedhom #blcsm hubs.ly/Q041HfMF0
3
7
508
Ryan Chow retweeted

💫🌟🚨 New real-world evidence on EV dose reduction in advanced urothelial cancer🌟💫
🎓 @JAMAOnc study shows that starting treatment with reduced-dose enfortumab vedotin in 1L EV pembrolizumab:
🔵 Cuts treatment interruptions by ~50% (better tolerability)
🔵 Maintains overall survival — no compromise on outcomes
🔵 Benefits remain consistent in older & physiologically vulnerable patients
💡 Take-home: “Start low, go slow” may be a safe and practical strategy to keep patients on therapy without losing effectiveness.
jamanetwork.com/journals/jam…
@OncoAlert @myESMO @ASCO @BladderCaJrnl @MedicalwatchHQ
#BladderCancer #UrothelialCancer #OncoAlert #GUOncology #EV302



2
17
41
2,849
Ryan Chow retweeted

Jan 6
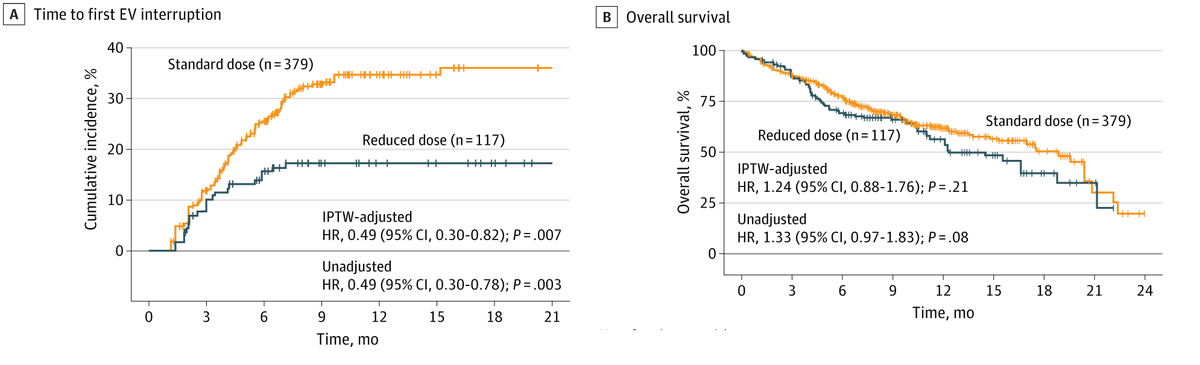
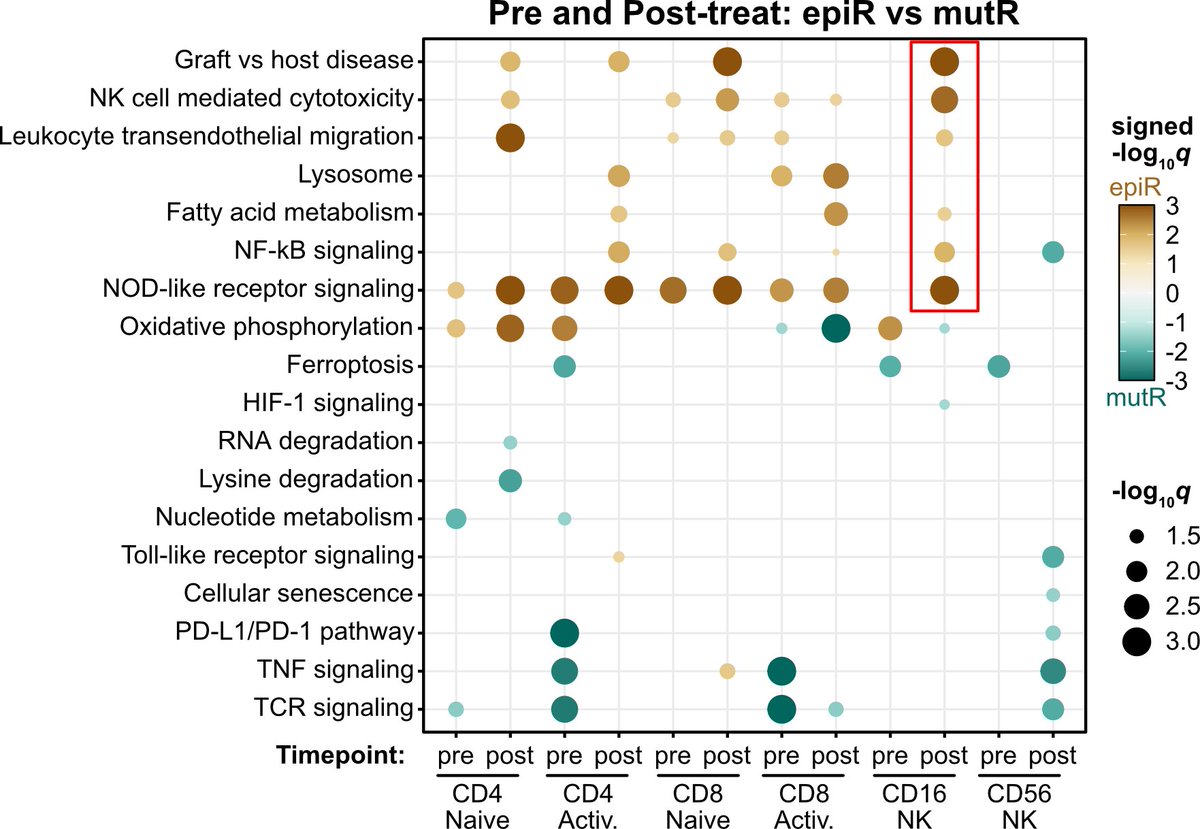
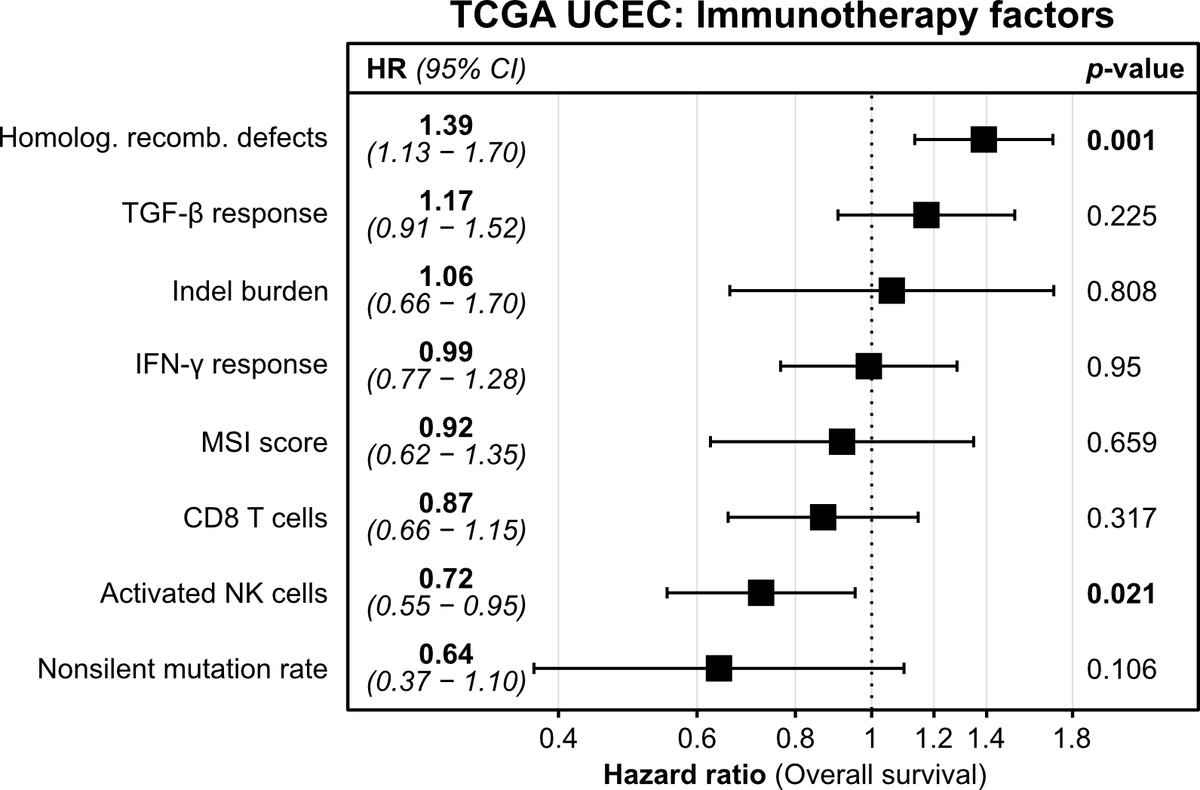
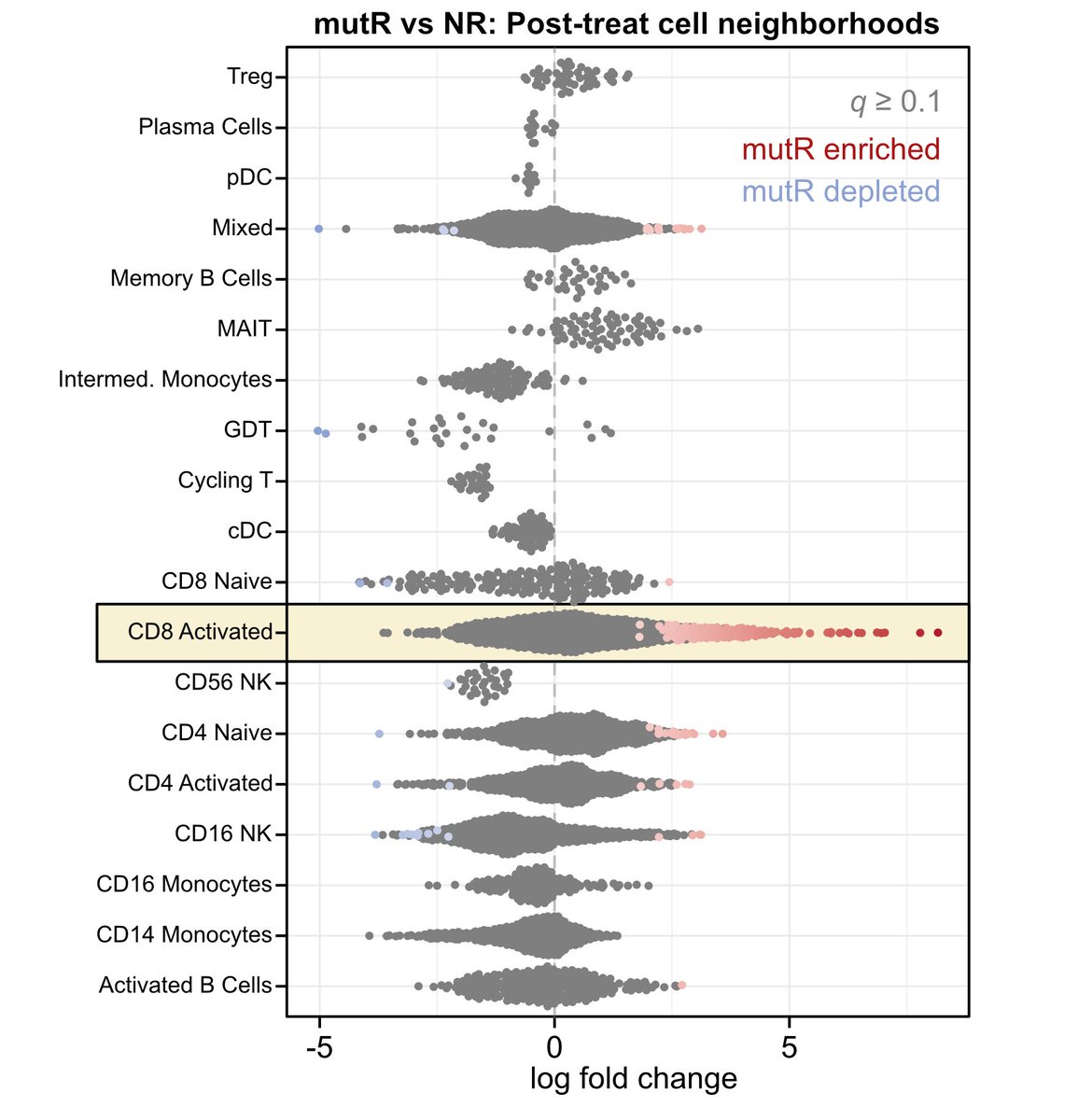
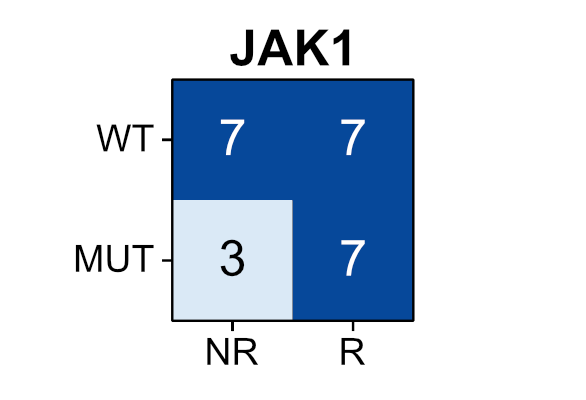
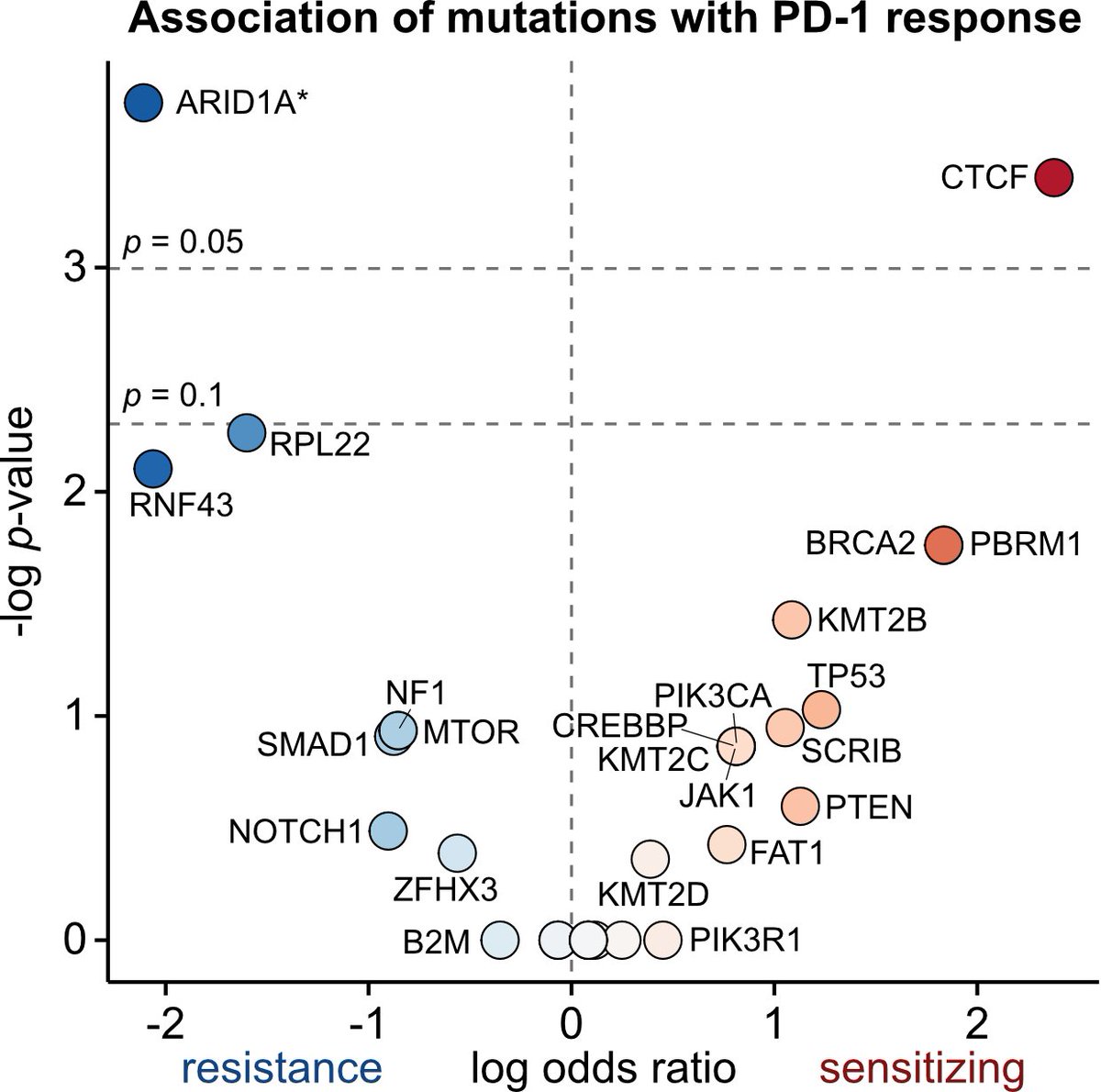
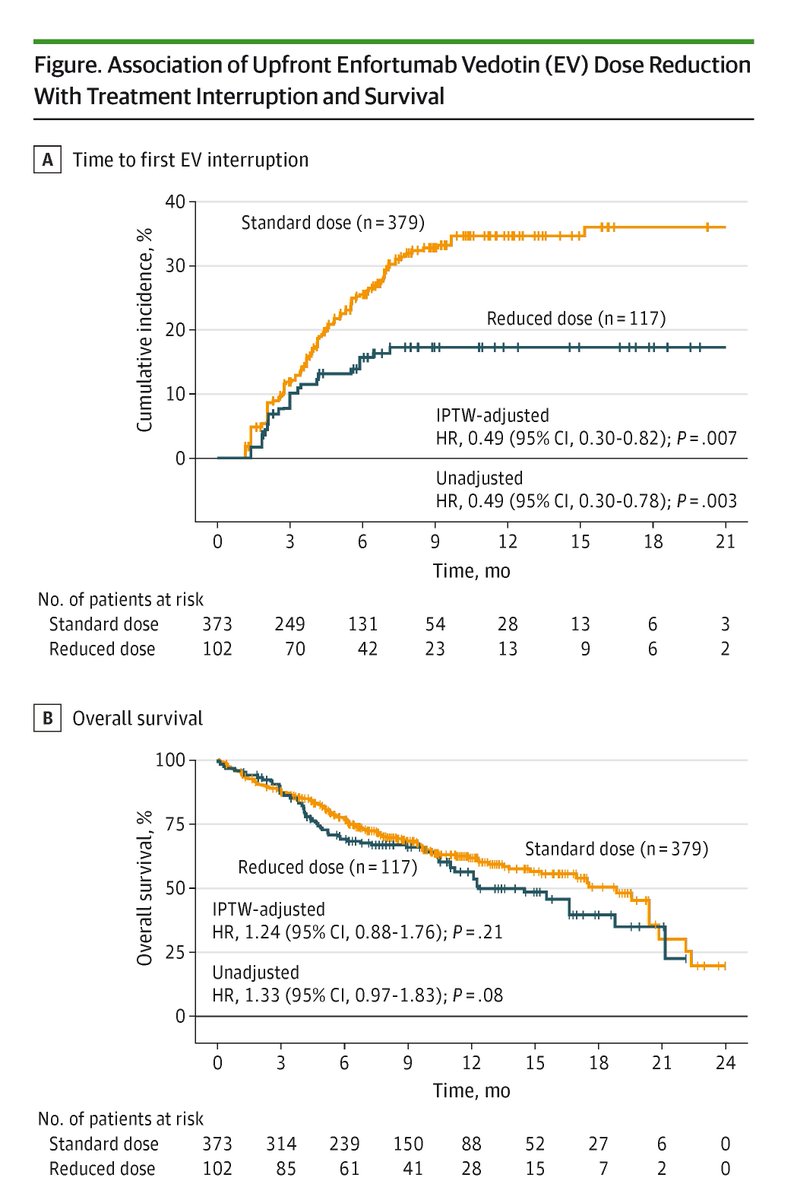
Among patients with advanced urothelial cancer, upfront enfortumab vedotin dose reduction was linked to a 50% reduction in treatment interruption risk but did not compromise overall survival.
ja.ma/3Lw71JH
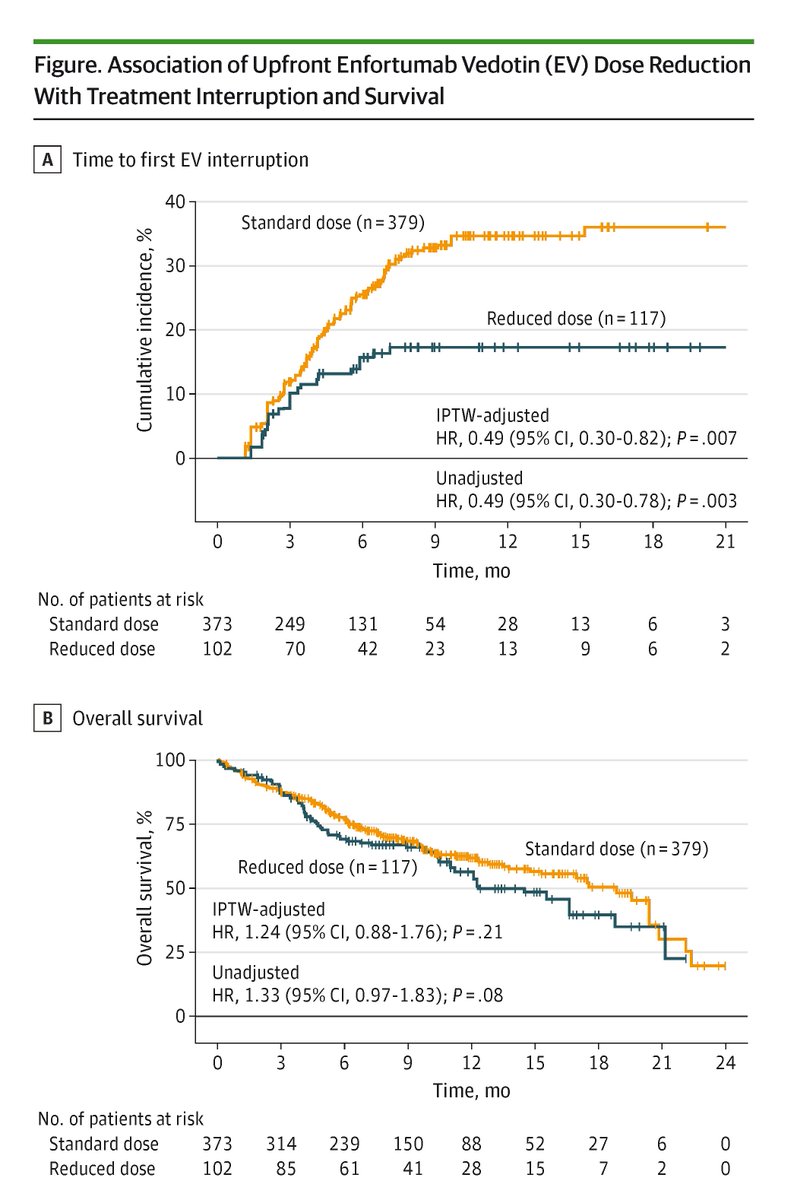
ALT Figure shows association of Enfortumab Vedotin dose reduction with treatment interruption and survival. Graph A plots time to first EV interruption. Graph B plots overall survival. Standard dose, reduced dose, and the number of patients at risk are shown.
20
68
6,261
Ryan Chow retweeted

17 Dec 2025
A retrospective cohort study led by @ScienceChow & team supports reducing the dose of the antibody-drug conjugate enfortumab vedotin when patients receive it as first-line therapy pembrolizumab for the most common form of bladder cancer. @ASCOPost spr.ly/60157h5QH
2
4
683
Ryan Chow retweeted

12 Dec 2025
New study in #UrothelialCancer shows upfront lower-dose enfortumab vedotin (EV) pembrolizumab:
🔻 Lowered risk of EV interruption (HR = 0.49)
⏱️ Similar OS vs standard-dose EV
💡 Efficacy similar in vulnerable pts (80 , poor performance)
🔹@ScienceChow
ascopost.com/news/december-2…

4
5
619
Ryan Chow retweeted

16 Nov 2025
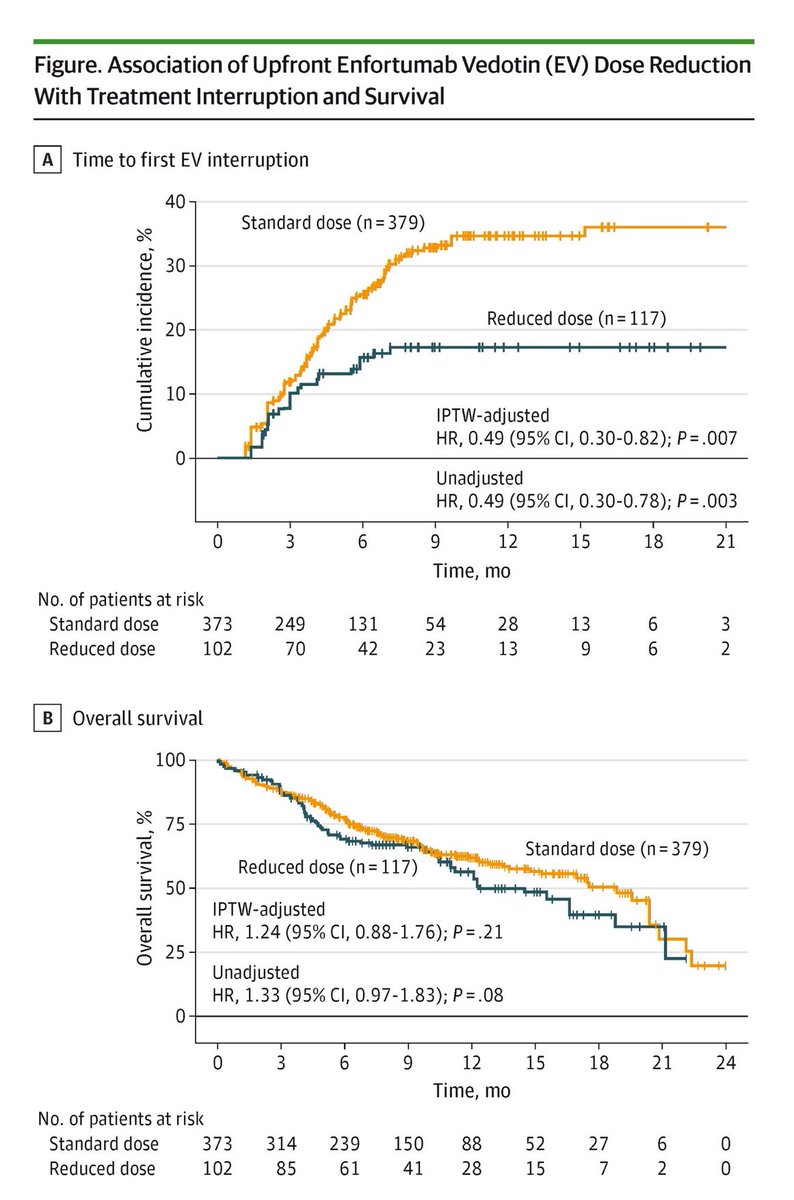
Early EV dose reduction in advanced urothelial cancer ↘️ treatment interruption risk by half, yet survival stayed the same.
@JAMAOnc @OncoAlert @WorldBladderCan @Uromigos
jamanetwork.com/journals/jam…
3
37
89
10,002
Ryan Chow retweeted
15 Nov 2025
Among patients with advanced urothelial cancer, upfront enfortumab vedotin dose reduction was linked to a 50% reduction in treatment interruption risk but did not compromise overall survival. ja.ma/47L9DdG
3
32
93
20,093
Ryan Chow retweeted

13 Nov 2025
Today in @JAMAOnc, we share new real-world evidence supporting reduced-dose EV Pembro for 1st-line rx of advanced urothelial cancer (a dz of older adults) led by @PennCancer fellow @ScienceChow & @Ron_cology
2
6
17
2,534
Ryan Chow retweeted

7 Aug 2025
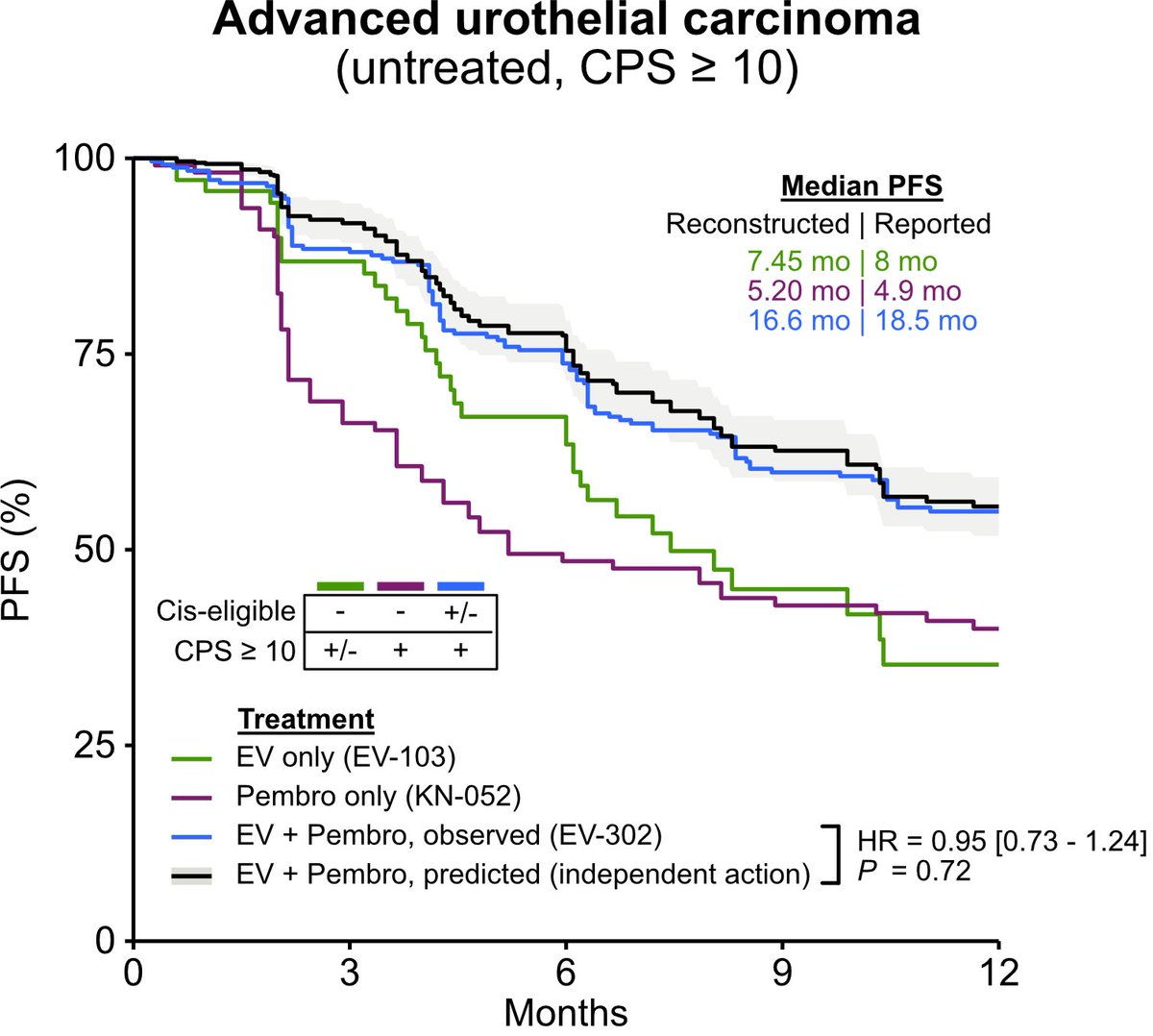
Is there a synergistic effect between enfortumab vedotin (EV) and pembrolizumab (P) for untreated advanced urothelial carcinoma in clinical setting?
This analysis suggests no!
EV P seems to follow a an independent drug action model with no evidence of synergy

7
22
102
12,373

6 Aug 2025
Enfortumab vedotin pembrolizumab (EV P) has revolutionized how we treat urothelial cancer. @Ron_cology and I were curious: is the efficacy of EV P better explained by synergistic or independent drug action? New in @UrolOncol: doi.org/10.1016/j.urolonc.20… (1/4)
4
2
7
261
6 Aug 2025
In an exploratory analysis, we also evaluated patients with PD-L1 CPS >10, who are more likely to respond to pembro. In this biomarker-selected group, observed PFS for EV-302 was again well-explained by independent activity of EV and pembro. (3/4)
1
63
6 Aug 2025
These findings suggest that distinct patient subgroups respond to EV vs pembro, as opposed to an emergent synergistic effect with combination EV P. Important implications for drug development in this setting! @PennCancer (4/4)
1
52
Ryan Chow retweeted

16 Jan 2025
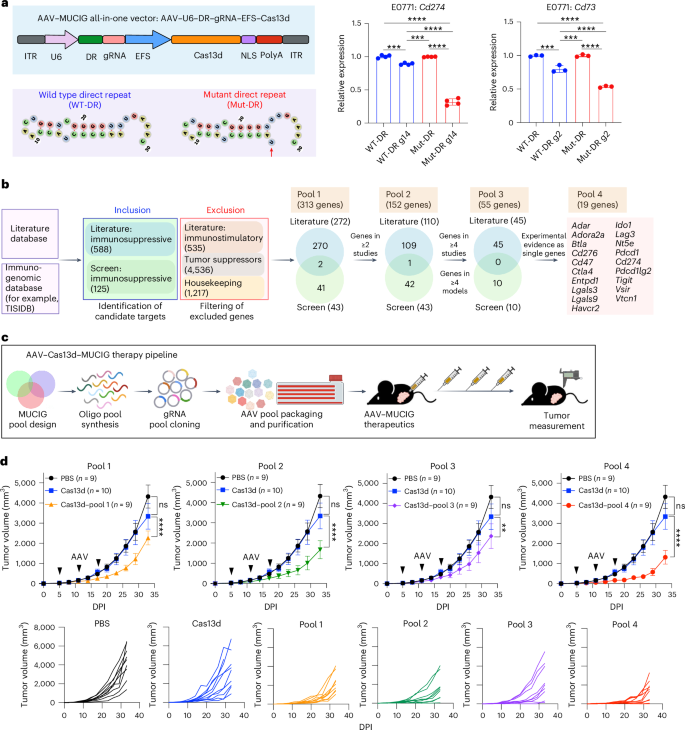
Multiplexed inhibition of immunosuppressive genes with Cas13d for combinatorial cancer immunotherapy go.nature.com/4gYcS4Z
8
30
113
13,975
Ryan Chow retweeted

21 Oct 2024
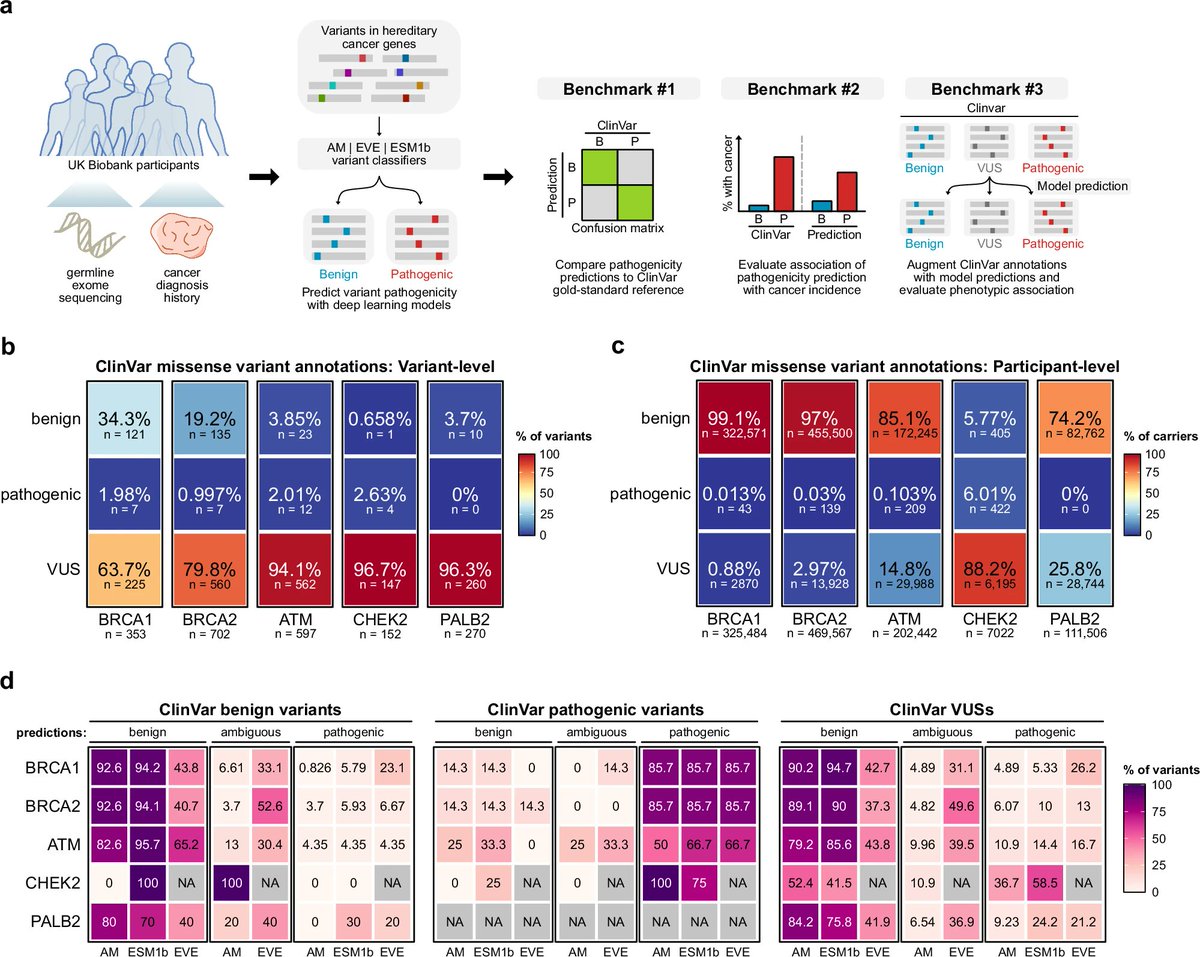
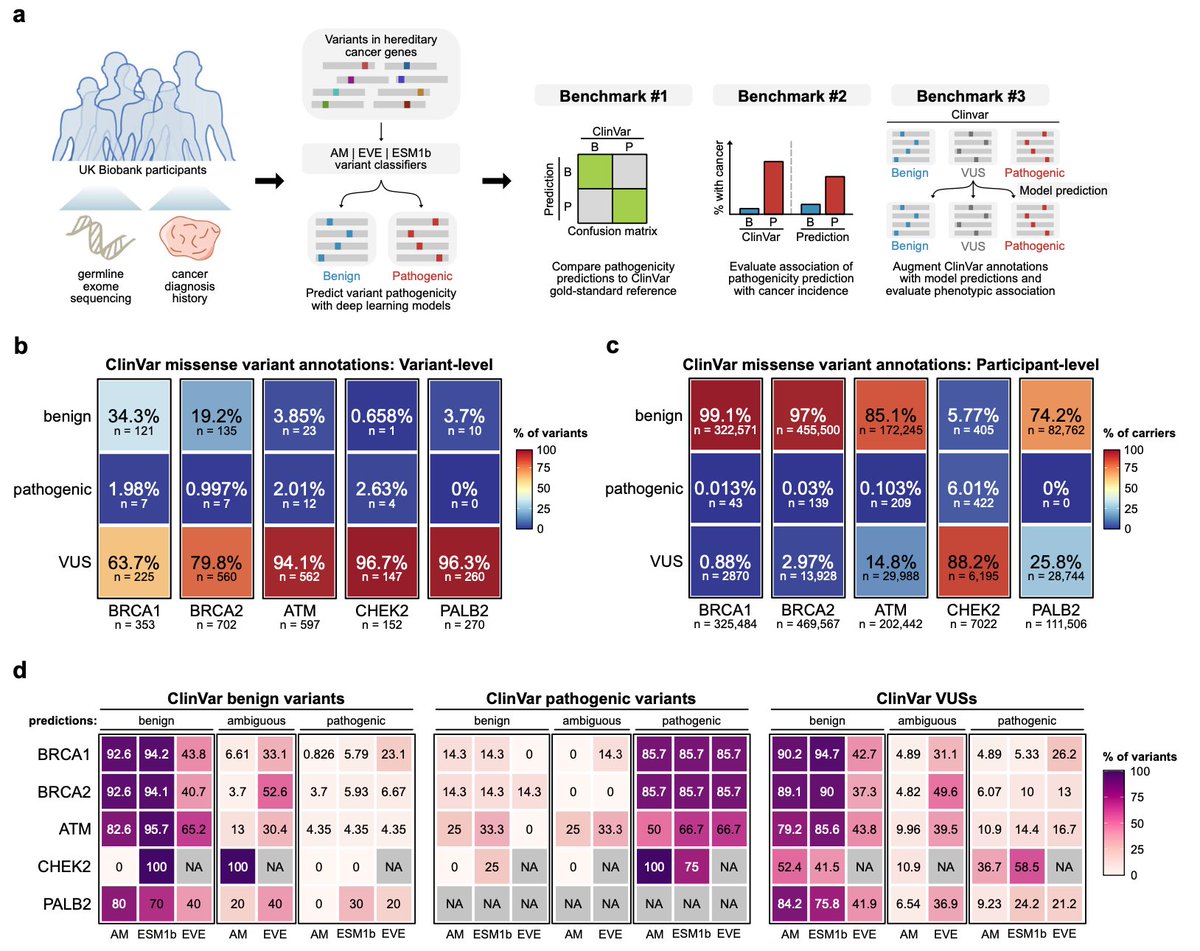
New @Nature_NPJ paper by @ScienceChow co-mentored w/ @KLNathanson @BasserBRCA on reliability of #deeplearning to predict variant pathogenicity. #AI recapitulate ClinVar classifications for pathogenic variants, but poorly predict pathogenicity for VUS's #breastcancer
21 Oct 2024

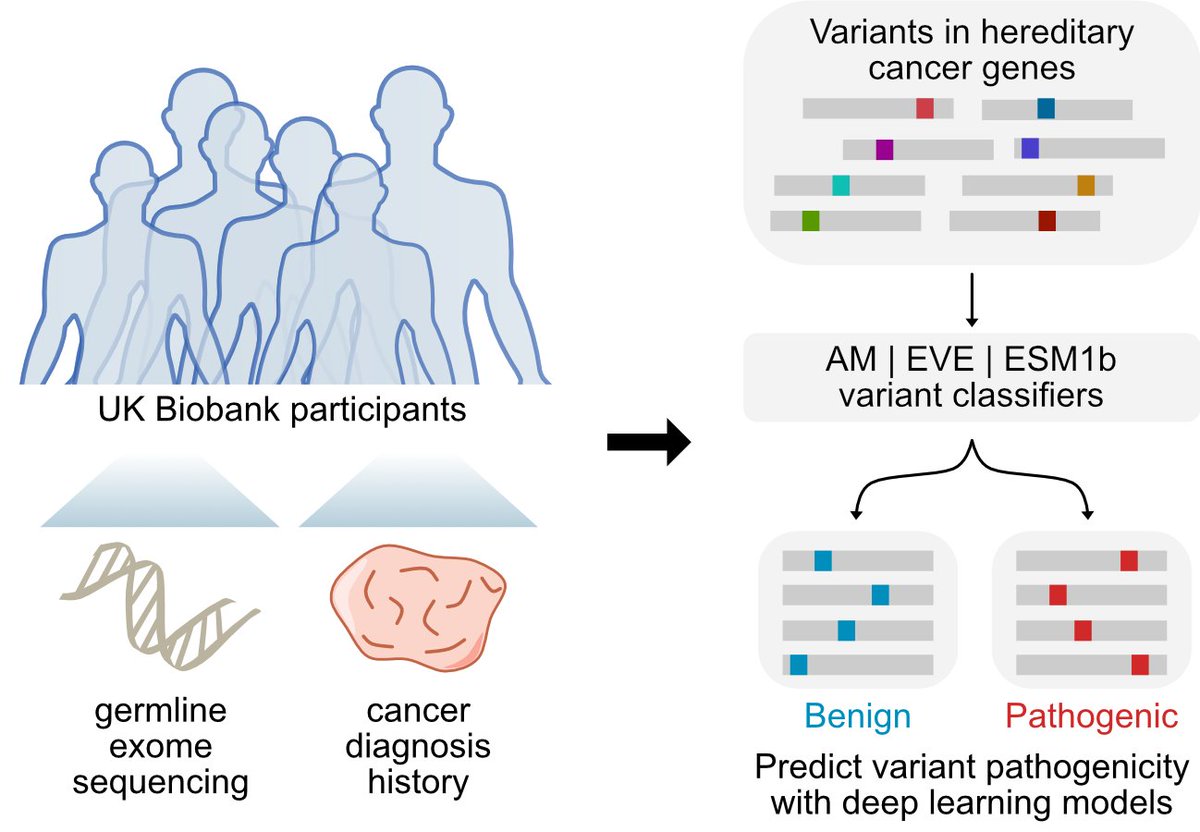
#DeepLearning models have been developed to predict missense variant pathogenicity -- but how well do these models perform in a real-world clinical setting? Thrilled to share our latest in npj Precision Oncology! @Nature_NPJ | nature.com/articles/s41698-0… (1/8)
3
18
3,984
Ryan Chow retweeted

20 Oct 2024
Phenotypic Evaluation of Deep Learning Models for Classifying Germline Variant Pathogenicity @Nature_NPJ
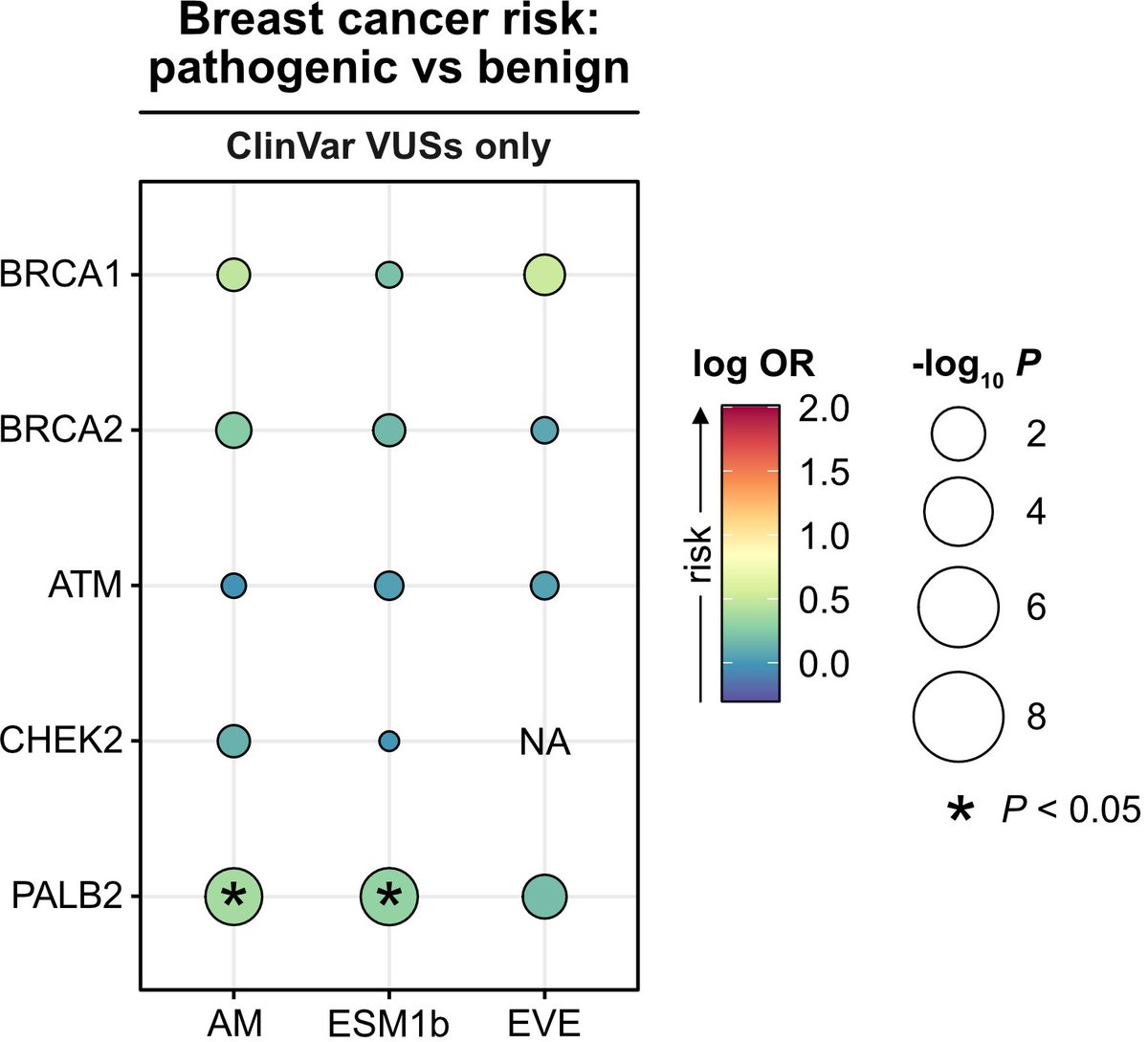
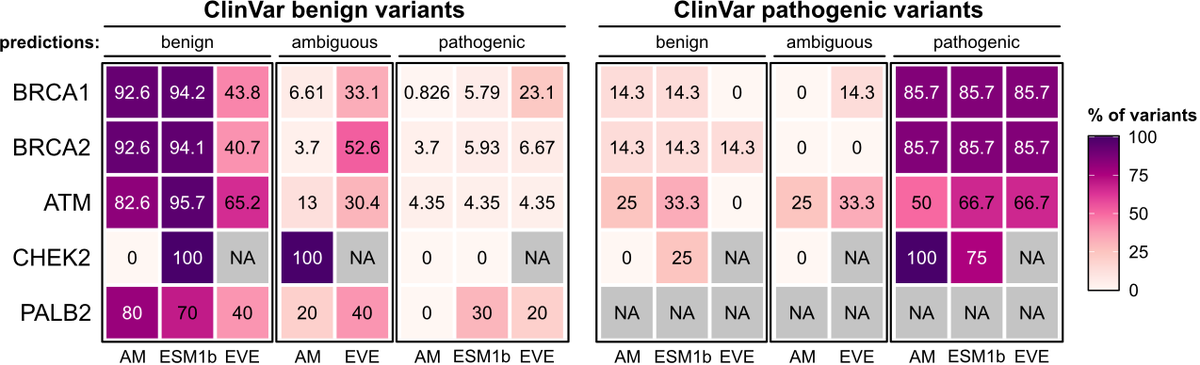
• This study evaluates the real-world utility of three state-of-the-art deep learning models—AlphaMissense, EVE, and ESM1b—in classifying germline variants associated with hereditary cancer risks.
• Using data from 469,623 UK Biobank participants, the study focuses on missense variants in key cancer-related genes, including BRCA1, BRCA2, ATM, CHEK2, and PALB2.
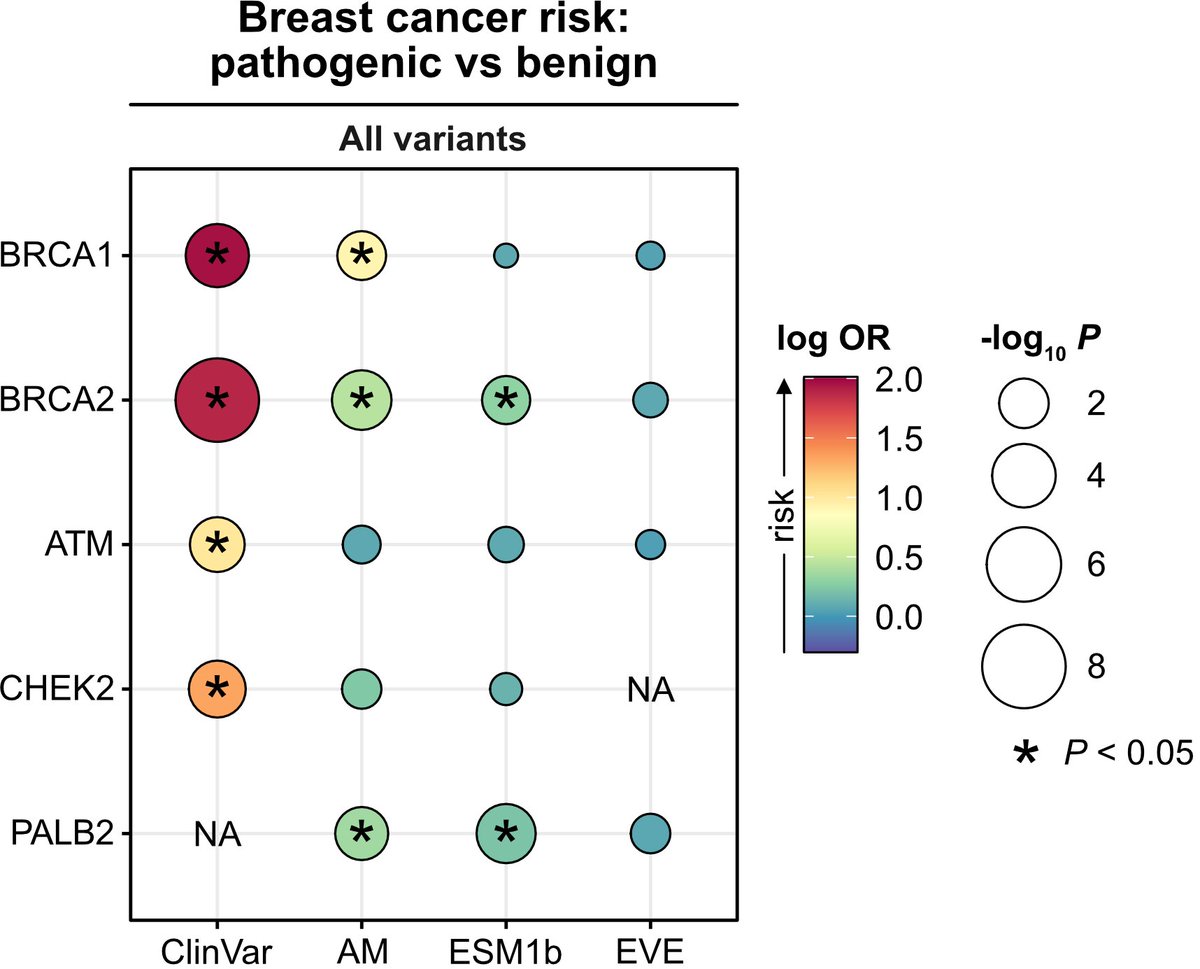
• AlphaMissense and ESM1b models were able to identify pathogenic BRCA1 and BRCA2 variants that conferred increased risk for breast and ovarian cancer, but they struggled with certain other genes like ATM and CHEK2.
• Notably, AlphaMissense identified potentially pathogenic PALB2 variants, which were previously categorized as variants of uncertain significance (VUS) by ClinVar, hinting at the models’ potential for refining variant classification.
• Despite their success with some genes, all models exhibited limited accuracy in distinguishing VUSs associated with increased cancer risk, underscoring the need for cautious interpretation in clinical practice.
• Composite classifiers that combined ClinVar annotations with deep learning predictions reduced the proportion of participants classified as VUS carriers, though at the cost of predictive power.
• The study highlights the importance of gene-specific thresholds, as a uniform cutoff reduced model accuracy across multiple genes, indicating that custom thresholds may improve performance.
• The authors emphasize the need for diverse genomic data to mitigate biases in current models, especially since VUSs are more prevalent in non-European populations.
• While the study shows promise for integrating deep learning in clinical settings, it concludes that deep learning models are not yet ready to fully replace traditional variant classification methods for clinical decision-making.
@ravi_b_parikh @KLNathanson @ScienceChow
💻Code: github.com/rdchow/UKB_pathog…
📜Paper: nature.com/articles/s41698-0…
2
11
1,352
21 Oct 2024
#DeepLearning models have been developed to predict missense variant pathogenicity -- but how well do these models perform in a real-world clinical setting? Thrilled to share our latest in npj Precision Oncology! @Nature_NPJ | nature.com/articles/s41698-0… (1/8)
1
5
2,926
21 Oct 2024
The notable exception: PALB2. Considering the sparse/conflicting ClinVar annotations for PALB2, this represents a concrete example where current #DeepLearning models could already inform variant classification and thus impact clinical decision making. (7/8)
1
130
21 Oct 2024
Thanks for reading! Open-access at nature.com/articles/s41698-0…. This study would not be possible without all the @uk_biobank participants. Many thanks to @KLNathanson & @ravi_b_parikh for their mentorship! @PennCancer @PennMedicine @PC3Innovation @PennIMResidents (8/8)
1
143