
Joined September 2022
- Tweets 4,937
- Following 144
- Followers 113
- Likes 16,198
102 Photos and videos










Pinned Tweet

6 Nov 2023
My top ten favorite books meaning I think about them at least every few days:
Foundation
Rendezvous With Rama
Midnight at the Well of Souls
Little Big
The Arabian Nightmare
The Maltese Falcon
Solomon's Vineyard
My Family and Other Animals
The French Lieutenant's Woman
Voss
1
14
6,651
Tyler Moore retweeted

May 20
New blog!
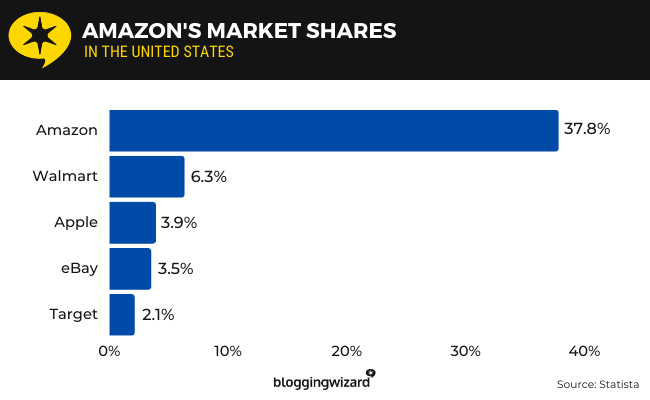
Synthetic Persona Pretraining (SPP): Alignment from Token Zero
Current alignment is shallow - values bolted on after pretraining can be routed around. To solve this, we wrote the desired persona directly into pretraining data. Early results, but we're very excited. 🧵
17
40
302
46,707
Tyler Moore retweeted

May 22
What if an AI system could sketch research directions before the field converges on them?
This paper explores the “alien space” of science: coherent ideas that are unlikely to be cognitively available to today’s research community.
arxiv.org/abs/2603.01092


(Current) LLM-based ideation is biased toward what the field already finds easy to think.
We formalize this bias as cognitive availability and use it to identify coherent but overlooked research directions: The Alien Space of Science. 🧵
arxiv.org/abs/2603.01092

3
17
97
16,616
Tyler Moore retweeted

Feb 12
"Pros won’t use generative AI, and when the bubble pops, nobody will ever talk about it again."
No. That’s delusional.
1/ Generative AI is already being used professionally at the level of big studios like Disney ($1B to OpenAI), and there’s zero doubt that studios like Industrial Light & Magic, Netflix, Hollywood VFX experts, etc. are already experimenting with it too.
Or do you think they’re idiots? They’re not idiots at all. They have the experience and, more importantly, the DISTRIBUTION POWER.
The point is: someone with taste, judgment, and storytelling experience, basically from their living room, will have access to (almost, or not even almost) the same capability as the big guys, because the pure "making stuff" skills have been commoditized, and the new way to create is just NATURAL LANGUAGE.
What hasn’t been commoditized is good taste, the ability to create great stories that move people, and the ability to get them in front of people.
So in the end, what wins is story quality and distribution.
Having good taste, making a name for yourself, and owning strong IP (Marvel, etc.) will still matter.
That’ll be true right up until AI is genuinely opinionated and can create by itself: if it comes to that, with zero human direction, stuff as good as (or better than) the very best human experts today, and on top of that, interactive in real time... Because yeah: there’s nothing in this universe that actually prevents that from happening.
BUT WE’RE NOT THERE. For now, generative AI is a tool that needs direction and taste to make anything decent. And I hope it stays that way for a long time, because otherwise that’s going to be a brutal hit to humanity’s ego.
2/ On the "bubble": you have to distinguish between a stock valuation bubble (possible, I actually believe it) vs a bubble like some people imagine where it "pops" and we never hear about AI again. That obviously makes no sense given how insanely useful it is. It can only grow, and it’s going to grow fast, regardless of any stock market drawdowns (the internet kept growing even when valuations got nuked in 2000).
Either way, the near future is going to be extremely interesting.
68
44
375
75,190
Tyler Moore retweeted

Almost no one has articulated a positive vision for what comes after superintelligence. What should we be trying to aim for?
Utopias from history look clearly dystopian to us, and we should expect the same for our own attempts. We don’t know enough to know what utopia looks like.
The main alternative framework is “protopianism”: solving the most urgent problems one by one, not guided by any big-picture view of society’s long-run course. I prefer protopianism to utopianism, but it gives up too much. The transition to superintelligence will present many problems all at once, and may need to choose between very different solutions to the same problems. We need a way to prioritise and plan.
So I want to introduce a third framing: viatopia.
A viatopia is a state of society that is *on track* for a near-best future, whatever that might look like. A teenager might not know what they want to do with their life, but know that a good education keeps their options open.
My current guesses for what viatopia looks like: material abundance, technological progress, coordination to avoid conflict, low catastrophic risk—plus preserving society-wide optionality, cultivating reflection, and structuring deliberation so better ideas win out.
I have a new essay on this idea — link in reply.

43
41
362
42,196
Tyler Moore retweeted

4 Dec 2025
2. Cell Reprogramming
It won't work. Cellular reprogramming includes “pluripotent reprogramming,” “partial reprogramming,” and “direct reprogramming.” In pluripotent reprogramming, somatic cells are completely reverted to a pluripotent state; in partial reprogramming, these cells may be rejuvenated but do not lose their cellular identity; and in direct reprogramming (transdifferentiation), somatic cells are directly converted into another type of differentiated cell.
The iPS cells generated by pluripotent reprogramming, according to the “Telomere DNA and Ribosomal DNA co-regulation model for cell senescence,” have been found to have significantly increased telomere length and rDNA array length compared to senescent somatic cells, indicating that the cellular age is reversed to 0 years. However, iPS cells or adult cells differentiated from iPS cells can also be subject to immune rejection. For example, on May 13, 2011, Nature reported that American scientists transplanted mouse iPS cells or adult cells differentiated from iPS cells into mice from which the cells were derived. It was originally thought that there would be no rejection due to the identical genetic background, but the mice rapidly rejected the transplanted cells [1]. On August 11, 2022, a paper published in Nature Genetics confirmed that nearly three-quarters of iPS cell lines have severely damaged DNA. Moreover, allogeneic iPS cells with the same homozygous human leukocyte antigen (HLA) haplotype as the patient can still cause immune rejection [2], indicating that even with the same HLA, allogeneic stem cells will still be rejected. In 2024, Deng Hongkui and others published a paper in Cell on the transplantation of islets derived from chemically induced pluripotent stem cells (CiPSCs) for the treatment of type 1 diabetes. The use of immunosuppressive agents indicates that small molecule reprogramming can also be rejected [3]. The original text is as follows: Before CiPSC-islets transplantation, the patient was maintained on immunosuppressive therapy with tacrolimus (2-2.5 mg/day), mycophenolate mofetil (1 g/day) and methylprednisolone tablets (8 mg/day) due to liver transplantation. For CiPSC-islets transplantation, Basiliximab (20 mg) was used for induction therapy on day 0 and day 4. Etanercept was administrated intravenously on day 0 (50 mg) and subcutaneously on day 3, day 7 and day 10 (25 mg) to alleviate inflammatory reactions. For maintenance therapy, the previously described immunosuppressive maintenance regimen for liver transplantation was continued. Cefazolin Sodium was administered for infection prophylaxis during the perioperative period.
Partial reprogramming is also not viable because it has been found that the telomere length does not increase or even slightly decreases in partial reprogramming [4], and it is estimated that the length of the rDNA array does not increase either. After the cessation of Yamanaka factor expression, the epigenetic age quickly reverts to the previous state, and aging symptoms accumulate rapidly. Multiple cycles of treatment only extended the lifespan of progeroid mice by 30%, but failed to extend the lifespan of wild-type mice, indicating that partial reprogramming did not reverse the underlying mechanisms of aging. If in vivo reprogramming is excessive, it can cause mouse death or the formation of iPS cells and fatal teratomas derived from iPS cells. Partial reprogramming uses doxycycline, which is mentioned below to significantly extend the lifespan of Caenorhabditis elegans and mice. The earliest in vivo partial reprogramming experiments were conducted in 2016, and to date, they have not extended the lifespan of normally aging mice. In 2016, Cell reported that cyclic transient expression of the four Yamanaka factors—Oct4, Sox2, Klf4, and c-Myc (abbreviated as OSKM)—in mice significantly extended the lifespan of progeroid mice, but did not extend the lifespan when OSKM was induced in normally aging mice (12 months old) [5]. Altos Labs, founded with $3 billion in investment from the world's richest man Jeff Bezos and Yuri Milner, reported in September 2024 that partial reprogramming only increased the median lifespan of wild-type mice by 12%, which is not as effective as small molecule anti-aging drugs. It can be said that the path of partial reprogramming has been declared a failure [6].
Direct reprogramming has not been found to reverse the aging phenotype [7].
[1] Zhao, T., Zhang, ZN., et al. (2011). Immunogenicity of induced pluripotent stem cells. Nature, 474, 212–215.
[2] Ozaki, M., Iwanami, A., et al. (2017). Evaluation of the immunogenicity of human iPS cell-derived neural stem/progenitor cells in vitro. Stem Cell Res., 19, 128–138.
[3] Wang, S., et al. (2024). Transplantation of chemically induced pluripotent stem-cell-derived islets under abdominal anterior rectus sheath in a type 1 diabetes patient. Cell, 187(22), 6152 - 6164.e18.
[4] Gill, D., Parry, A., et al. (2022). Multi-omic rejuvenation of human cells by maturation phase transient reprogramming. eLife, 11:e71624.
[5] Ocampo, A., Reddy, P., et al. (2016). In Vivo Amelioration of Age-Associated Hallmarks by Partial Reprogramming. Cell, 167, 1719-1733 e1712.
[6] Sahu, S. K., et al. (2024). Targeted partial reprogramming of age-associated cell states improves markers of health in mouse models of aging. Sci Transl Med.
[7] Mertens J, Paquola ACM, et al. (2015). Directly Reprogrammed Human Neurons Retain Aging-Associated Transcriptomic Signatures and Reveal Age-Related Nucleocytoplasmic Defects. Cell Stem Cell. 17:705-718.
[8] Huang, Bilu. (2025). The Information Theory of Aging Is Not Reliable. Available at SSRN
2 Dec 2025

New article: The Information Theory of Aging (ITOA) states that epigenetic drift is a cause of aging, with cells taking up new identities. New review says slowing a process called "mesenchymal drift" has emerged as is a new strategy for rejuvenation
7
9
87
8,555
Tyler Moore retweeted

13 Nov 2025
very cool paper - tl;dr it's possible to steer models by taking a weight difference rather than an activation difference. arxiv.org/abs/2511.05408
7
33
279
29,093
Tyler Moore retweeted

13 Nov 2025
In five hours this has only gotten 31 likes and about 50 more from my reshare.
Nuts how hard it is to get reach here on X.
Everyone is missing how big a deal this is.
It introduces deep physics understanding into world models.
Google and World Labs sucked the oxygen out of the room.
Turn on audio.

Introducing PAN — MBZUAI’s New World Model for Interactive Intelligence
Developed by MBZUAI’s Institute of Foundation Models, PAN is built for simulation, prediction, and agentic reasoning.
Unlike traditional video generators that only output frames, PAN maintains a persistent internal state that evolves when guided with natural language.
Its Generative Latent Prediction architecture combines:
• A latent encoder to capture the world state
• A dynamics module that evolves that state step-by-step
• A video diffusion decoder that visualizes outcomes
By decoding at every step using a causal sliding-window diffusion process, PAN stays grounded in real-world physics and maintains long-horizon continuity, a leap beyond single-shot models.
Evaluated on action fidelity, long-horizon stability, and simulative planning, PAN delivers state-of-the-art performance compared to open models and rivals leading commercial systems.
For robotics, autonomy, and decision support, PAN is a foundation for the next wave of intelligent, foresight-driven AI.
panworld.ai
36
44
422
57,460
Tyler Moore retweeted

28 Oct 2025
Great work from @SmithaMilli, @MicahCarroll, and team.
Smitha keeps putting out bangers that have immediate application but are also address longer-term alignment concerns.
27 Oct 2025

can we finally use natural language to optimize for deeper notions of what users want from their recommender systems?
3
2
13
2,380
Tyler Moore retweeted
9 Oct 2025
Unified Theory of Aging - Why Do We Age?The Telomere DNA and Ribosomal DNA Co-regulation Model for Cell Senescence (TRCS)
Bilu Huang
Available at SSRN: ssrn.com/abstract=5558378
Abstract
When killifish species with different lifespans that evolved under distinct rainy-season lengths are raised together in the same aquarium, their lifespan differences persist-evidence that aging after sexual maturation is still genetically programmed. Individual aging is driven by replicative senescence of adult stem cells. Our work shows that cellular senescence is co-regulated by telomeres and ribosomal DNA (rDNA) through the P53 pathway. Moreover, the 11 other acknowledged hallmarks of aging are also downstream consequences mediated by telomere and rDNA shortening through P53 pathway.
Keywords: Replicative senescence, Individual aging, Adult stem cells, Telomeres, rDNA, TRCS
If you can't explain it simply, you don't understand it well enough. —Albert Einstein
Aging and death are so frightening—so why do we age?
1. Aging is programmatically controlled, not the result of random damage accumulation.
When three killifish strains—one with a 3-month lifespan, one with a 9-month lifespan, and one with a 16-month lifespan—evolved under rainy seasons of corresponding lengths, are reared together in the same aquarium, their lifespan differences remain intact. This shows that post-maturational aging is still genetically programmed, because random damage accumulation cannot explain why three closely related, anatomically almost identical species differ so dramatically in longevity, nor why these differences match the duration of the rainy seasons in their native habitats [1].
2. Individual aging is caused by the replicative senescence of adult stem cells.
Replicative senescence is characterized by the phenomenon that with each cell division, daughter cells become older than the mother cell. Specifically, this involves the downregulation of overall protein and ATP synthesis rates, while the expression of some proteins is upregulated and others downregulated. This leads to a decline and alteration in cellular function, indicating that cellular senescence is also programmed, continuing until the cell ceases division upon reaching the Hayflick limit.
The somatic cells constituting the various tissues and organs of an individual are broadly classified into two categories: "adult stem cells" and "functional cells". Although no adult stem cells have been identified in the heart, cardiomyocytes can renew themselves through self-replication [2]. Therefore, cardiomyocytes serve as both adult stem cells and functional cells.
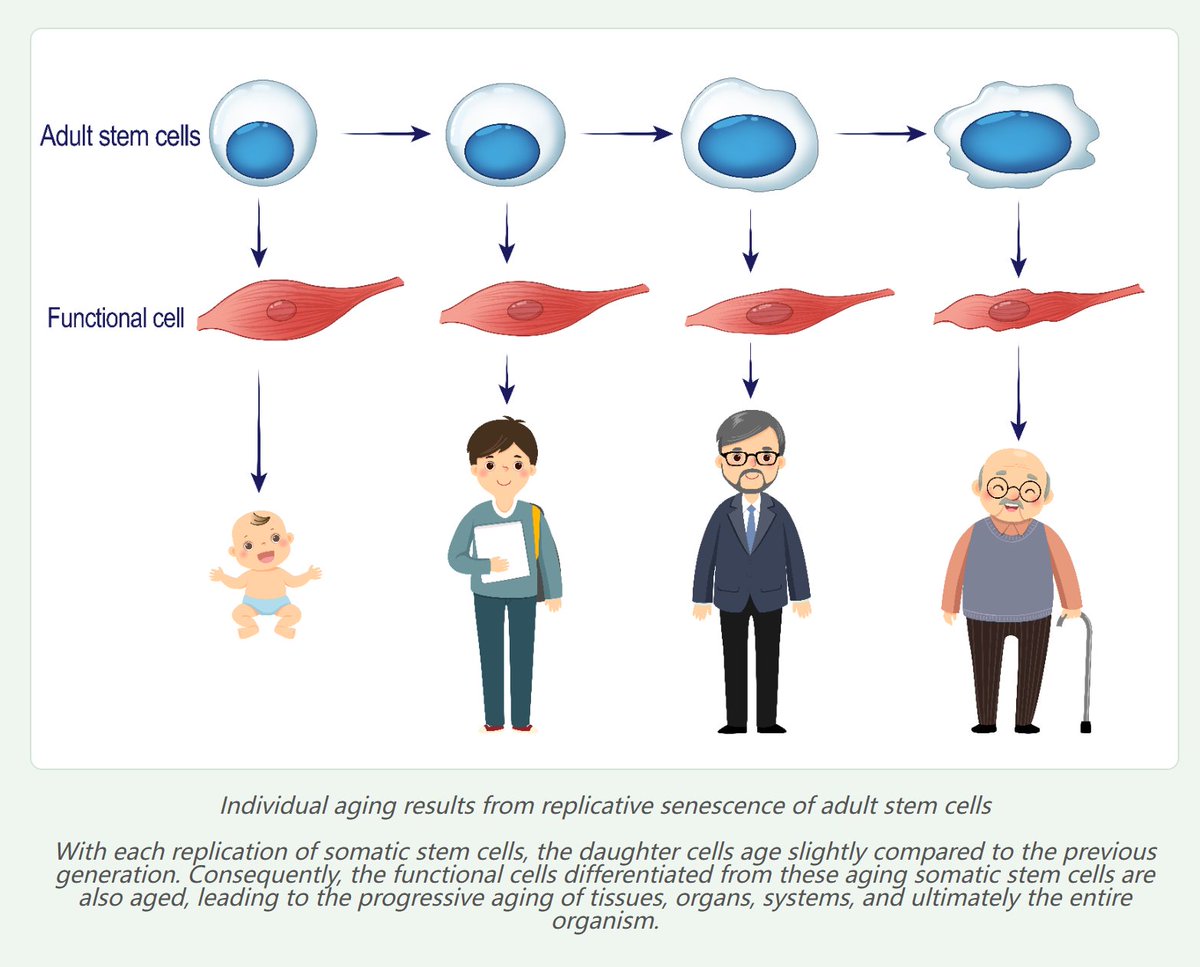
Both adult stem cells and functional cells can be cleared by the immune system due to factors such as cellular senescence, genetic mutations, or viral infections. They are then replenished through the self-replication and differentiation of adult stem cells. However, the adult stem cells themselves undergo replicative senescence due to repeated self-replication. Similarly, the functional cells differentiated from aged adult stem cells are also aged functional cells, leading to the gradual aging of tissues, organs, systems, and ultimately the entire individual (Figure 1).
For example, the adenohypophysis in the hypothalamus contains six types of somatic cells: somatotropes (secreting growth hormone), thyrotropes (secreting thyroid-stimulating hormone), corticotropes (secreting adrenocorticotropic hormone), gonadotropes (secreting gonadotropins), lactotropes (secreting prolactin), and a population of adenohypophyseal stem cells responsible for differentiating into these five hormone-secreting functional cell types. With advancing age, these adenohypophyseal stem cells gradually undergo replicative senescence. This results in the functional hormone-secreting cells derived from them becoming increasingly senescent, leading to age-related changes in the endocrine system.
Therefore, the fundamental cause of individual aging can be ultimately attributed to the replicative senescence of adult stem cells [3].
3. The replicative senescence of cells is co-regulated by telomeres and rDNA.
Telomeres are tandemly repeated multi-copy DNA arrays. Telomere shortening has been proven to act as a countdown timer for cell replication and senescence. However, in some cells where telomeres do not shorten, the number of cell replications and lifespan remain limited. Furthermore, it has been observed that maintaining telomere length with telomerase, while increasing the number of replications, ultimately does not prevent cells from ceasing replication and dying. Therefore, besides telomeres, there must be another multi-copy tandemly repeated DNA array in the nucleus that serves as a second countdown timer for cell replication and lifespan. Consequently, I proposed the "Telomere DNA and ribosomal DNA co-regulation model for cell senescence" (TRCS) [4]. This theory posits that the shortening of telomere and/or rDNA arrays leads to increased levels of the tumor suppressor protein P53, thereby inducing cellular senescence (Figure 2). Furthermore, the TRCS theory has been experimentally validated as correct, with rDNA contributing a greater weight to senescence than telomeres.
Since telomeres and rDNA are universal among all eukaryotic organisms, from yeast and nematodes to mice and humans, the mechanisms of aging across various species have been unified. 😊
Moreover, the 11 other acknowledged hallmarks of aging are also downstream consequences mediated by telomere and rDNA shortening through P53 pathway [1].
In summary, the ultimate cause of organismal aging is the progressive shortening of telomere and rDNA arrays in tissue-resident adult stem cells.
In theory, replacing the aged adult stem cells in aging tissues with young adult stem cells could rejuvenate the aged tissues. However, transplanted allogeneic stem cells will be cleared by the immune system. Transplanting autologous stem cells that have been expanded in vitro is also problematic, as the expansion process induces replicative senescence. Additionally, transplanting adult stem cells derived from induced pluripotent stem cells (iPSCs) generated from autologous cells is not viable, because these cells often harbor detectable DNA damage exceeding 70%, which leads to their immune clearance. Therefore, these approaches are not feasible. The only viable solution is to increase the length of telomeres and rDNA arrays in autologous adult stem cells [1]. Besides, interventions at the metabolic level and in signaling pathways can only slightly extend lifespan, come with significant side effects, and are unlikely to reverse aging.
Please follow, like, and share! Let's accelerate the quest to conquer aging. Challenge the death and race against time!
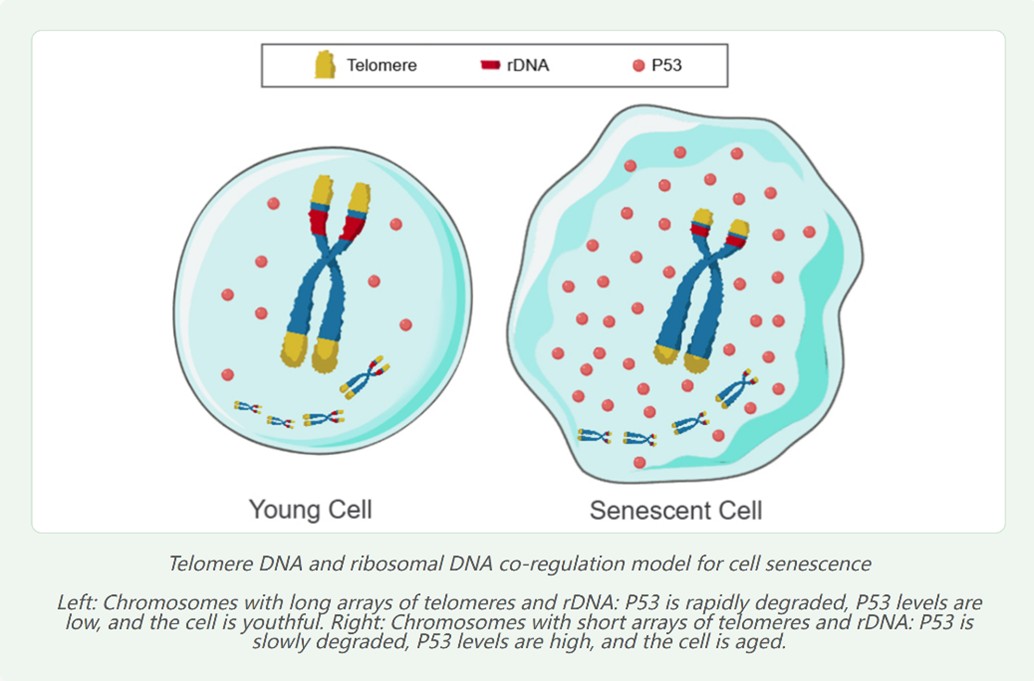

8 Oct 2025

We still don’t have a proper theory of aging.
That’s remarkable, given how far biology has come. Despite centuries of speculation and decades of data, there is still no unified, quantitative framework that explains how and why living systems age—and how we might stop it. What we have instead is a long list of partial explanations. By some counts, there are over 300 different theories of aging. That’s not a sign of success. It’s a sign of fragmentation.
But before building such a theory, we need to agree on aging phenomenology, decide what exactly needs to be explained. What are the core, undeniable features of aging that any model must capture? Here’s my proposal.
First, mortality doesn’t just increase with age—it increases exponentially. Across a wide range of species, from flies to humans, the probability of death doubles every few years of adult life. This is known as the Gompertz law, and it defines a central quantitative signature of aging. Deviations from this pattern are rare and interesting. Some short-lived animals flatten out their mortality risk late in life. Naked mole-rats barely age at all in demographic terms, even though their molecules do. Humans, meanwhile, keep on aging Gompertz-style even past the average lifespan.
What’s striking is how different this is from how machines fail. Most engineered systems don’t follow exponential mortality. They follow power laws. In mechanical systems—jet engines, turbines, bridges—the risk of failure usually increases according to a Weibull distribution: slow at first, then rapidly accelerating as wear accumulates. This is called “bath-tub shaped” failure. It's deterministic, often dominated by a single weak link.
Biology doesn’t work like that. It’s noisy, redundant, and dynamic. Cells talk to each other, systems compensate, and failures emerge from the collapse of coordination—not the snapping of one part. That’s why mortality in organisms rises exponentially, not deterministically. And that’s a big clue. It tells us aging isn’t just wear and tear. It’s something else—something emergent, statistical, and deeply biological.
Second, aging is fundamentally stochastic. Even genetically identical individuals in the same environment don’t die at the same time. They drift. And the spread of this drift—the variance of lifespan and age-related traits—actually increases with age. In humans, this variance seems to grow hyperbolically, as if all physiological systems are converging toward some kind of boundary near 120 years. The longer the species lives, the tighter this variance tends to be.
We see the same thing in the genome. DNA methylation clocks—those predictive models based on epigenetic patterns—derive most of their power not from deterministic changes, but from accumulating noise. Epigenetic drift turns out to be the main signal. And recent work shows that 70 to 90 percent of what these clocks “learn” is just statistical dispersion. The rest is the fine structure, the deviations from pure noise, that carry biological meaning.
Third, aging is scaled. Across mammals, bigger animals tend to live longer—and slower. Lifespan and developmental time follow predictable, quarter-power scaling with body size. Molecular damage, by contrast—mutations, methylation drift, oxidative stress—tends to scale inversely with lifespan. Bigger, longer-lived species accumulate this damage more slowly. There’s a deep geometry here, a kind of allometric constraint linking biology’s pace and its durability. A good theory of aging has to explain why.
Fourth, despite all the complexity, aging turns out to be low-dimensional. If you analyze enough biomarkers—genomic, proteomic, clinical—you find that most of the variation with age clusters along just a few directions. A few principal components can describe most of the story. Even organism-level performance metrics—frailty, lung function, heart rate ceiling—decline in a surprisingly linear way: roughly one percent per year, across the board. At the same time, individual differences grow. Aging is drift along a narrow ridge, with widening variance.
At last, let's remember that in practice nothing beats the effect of eating less. A good theory should give us a way beyond that. This is my proposal for what a theory of aging must explain. What’s yours?
As usual, mind following, like and repost - In fact, there is apparently only one way to explain all the listed features within a single theoretical framework (see the link to our recent work in the first comment)

21
60
258
34,006
Tyler Moore retweeted

4 Oct 2025
How can LLMs evolve continually in real-world industry without forgetting past tasks?
Enter: MoE-CL, a parameter-efficient adversarial mixture-of-experts framework for continual instruction tuning:
- Dedicated LoRA experts per task → preserve task knowledge
- Shared LoRA expert task-aware discriminator → transfer only task-relevant info
- Adversarial learning → balances retention & generalization
Tested on public & industrial benchmarks (incl. Tencent Video), MoE-CL cut manual review costs by 15.3%, proving scalable & practical for real-world deployment.
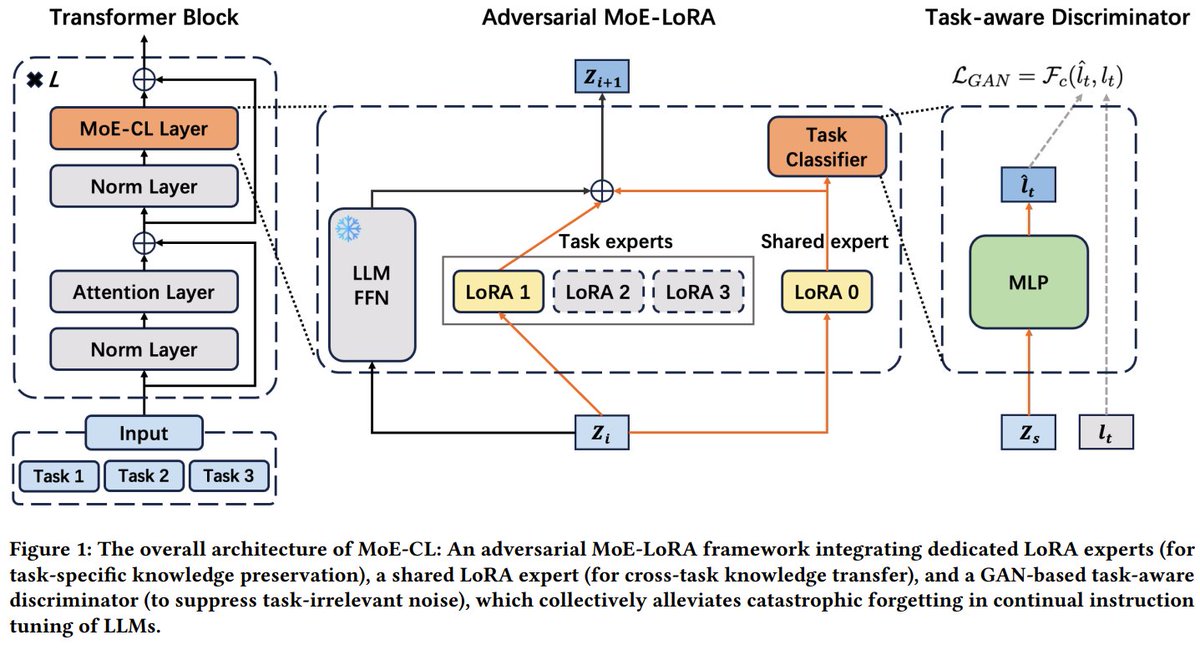
1
30
175
8,705
Tyler Moore retweeted

30 Sep 2025
Most LLMs learn to think only after pretraining—via SFT or RL. But what if they could learn to think during it? 🤔
Introducing RLP: Reinforcement Learning Pre-training—a verifier-free objective that teaches models to “think before predicting.”
🔥 Result: Massive reasoning boosts & gains that COMPOUND after post-training!
📝 Blog: research.nvidia.com/labs/adl…
🔗Paper: github.com/NVlabs/RLP/blob/m…
🧵↓
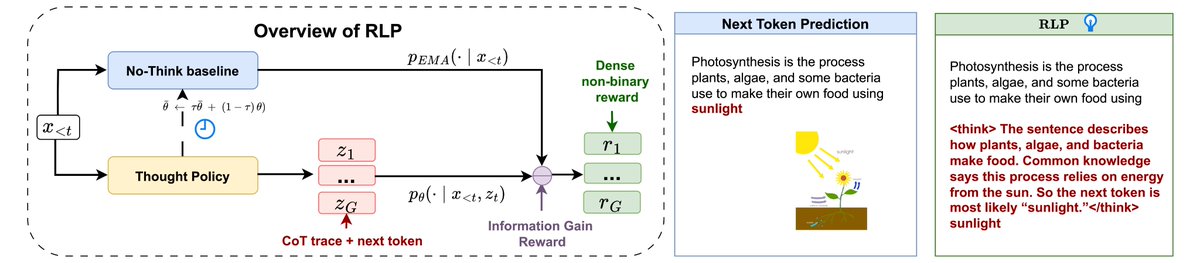
8
41
256
20,319
Tyler Moore retweeted

24 Sep 2025
"AI slop" seems to be everywhere, but what exactly makes text feel like slop?
In our new work (w/ @TuhinChakr, @dgolano, @byron_c_wallace) we provide a systematic attempt at measuring AI slop in text!
arxiv.org/abs/2509.19163
🧵 (1/7)


30 Jan 2025

help me fix get-4o slop
reply with examples of slop behavior
just a single sentence nothing crazy
what annoys you
what makes you wanna frisbee your laptop into a river
i'll respond to every comment
rt so we can maximize slop feedback
help me de-sloptimize our models
go
14
37
222
34,769
Tyler Moore retweeted
9 Sep 2025
The fear of death first hit me when I was 8. Since then, I have determined to bet on my life to conquer aging. My goal is to help humanity bid farewell to the cycle of the four seasons and usher in an eternal spring as soon as possible! I hope everyone cherishes life and can see the sun rise in the east every day. Therefore, my mission is not to just slow aging for longevity, but rejuvenation back to my most vigorous 30-year-old! Time flies away, relentless as the years; every news of death stirs in me a surge against aging. I will gather humanity’s most cutting-edge biotechnologies, develop theories of pinpoint precision, challenge the death and race against time!
After over 30 years of research, I have discovered the secret of cellular immortality. The reason why cells age is still under exploration globally. I have analyzed the various aging theories from around the world, and found that there were many loopholes and could not be justified. In order to uncover the mystery of cellular aging, I have proposed the Telomere DNA and Ribosomal DNA Co-regulation Model for Cell Senescence TRCS. This model has been validated through experiments.
Why do we age? Simply put, the various organs that make up an individual are composed of functional cells and adult stem cells. Functional cells constantly die for various reasons and are then replenished by adult stem cells through self-replication and cell differentiation to produce new adult stem cells and functional cells. However, each time an adult stem cell divides, it becomes older than the previous one, and the functional cells that differentiate from the aged adult stem cells are also aged cells, causing the organs and individuals to age further. Therefore, the key question is why do adult stem cells age? Our research shows that the degree of aging in adult stem cells is jointly regulated by the length of the telomere and rDNA array, and that rDNA plays a greater role in regulating cell aging than telomeres. Therefore, increasing the copy number of telomere DNA and rDNA in adult stem cells can reverse aging.
Through rigorous logical reasoning and experimental verification, we found the root cause of cellular senescence and Hayflick limits are telomere and rDNA. To surpass the limitations of lifespan, it is essential to extend telomeres and rDNA. Moreover, merely reversing epigenetic inheritance or modulating signaling pathways will prove ineffective.
The next step is to use this latest theory to develop biotechnology to rejuvenate aging adult stem cells and allow adult stem cells to divide indefinitely, which will be used to replace organ transplantation, cure various age-related diseases and greatly extend lifespan.
I don't speak English, so I used to only be able to publish papers in Chinese. Now, with the help of my team and AI tools, I started writing many English papers last year, aiming to address every major issue in the field of aging biology.
1. Aging Theory
1.1 Introduction to the Telomere DNA and Ribosomal DNA Co-regulation Model for Cell Senescence
Huang, Bilu. Available at SSRN: ssrn.com/abstract=5341657 or dx.doi.org/10.2139/ssrn.5341…
My hypothesis, the “Telomere DNA and Ribosomal DNA Co-regulation Model for Cell Senescence,” was initially published in Chinese in the journal Nagetive in 2021. Given the significance of this model in the study of aging research, I have revised the paper in English, incorporating preliminary in vitro cellular experimental validation results, in order to disseminate this innovative theory more widely.
1.2 Evidence and Experimental verification on “Telomere DNA and ribosomal DNA co-regulation model for cell senescence --TRCS ” (2021) -
x.com/biluhuang/status/19623…
1.3 FAQ: Telomere DNA and ribosomal DNA co-regulation model for cell senescence
lab.fuzhuangtx.com/en/faq
2. Feasibility Analysis of Other Aging Theories and interventions
I have not only proposed the "Telomere DNA and Ribosomal DNA Co-regulation Model for Cell Senescence" but also analyzed the feasibility of all existing aging theories and interventions. In fact, it was precisely because I scrutinized various aging theories and found that many of them had significant loopholes and could not be fully justified that I developed the "Telomere DNA and Ribosomal DNA Co-regulation Model for Cell Senescence." This model has been validated through experiments.
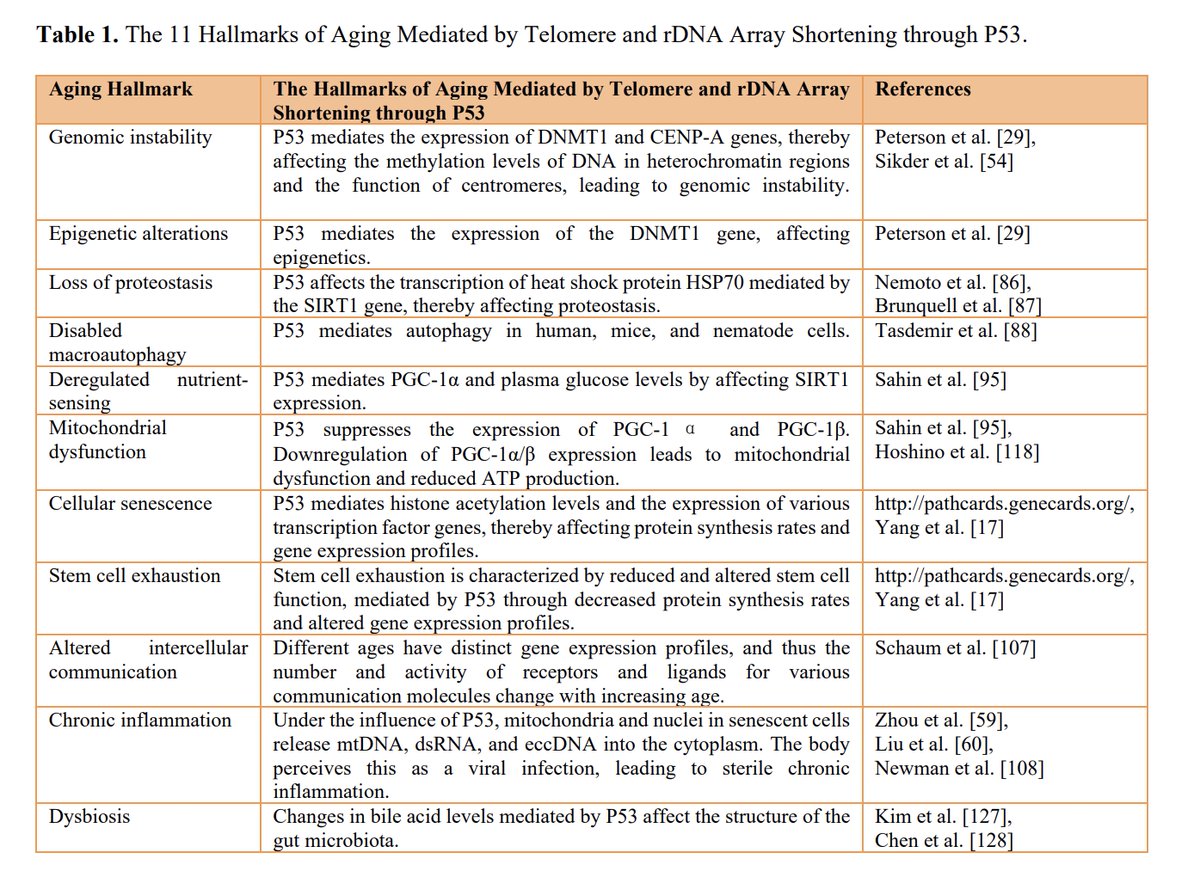
2.1 Causality of Aging Hallmarks
Bilu Huang , Xiaowen Hu. Causality of Aging Hallmarks. Aging and disease. 2025 doi.org/10.14336/AD.2025.054…
To solve the problem of aging, it is necessary to identify the root causes of aging. However, many hallmarks associated with aging have been observed and touted as the driving forces behind aging. In 2023, a review titled “Hallmarks of aging: An expanding universe” published in Cell listed the twelve major hallmarks of aging (recently it has grown to fourteen), but the authors did not provide causal arguments for each hallmark.
Interventions targeting the consequences of aging are not very effective, whereas only those targeting the causes can significantly extend lifespan. To clarify this issue, I established a causal relationship model for the 12 hallmarks of aging, asserting that among the twelve hallmarks of aging, only telomere shortening is the cause of aging and that other 11 hallmarks are merely downstreams mediated by telomere and rDNA arrays shortening through P53.
2.2 Demonstration of the Feasibility of Mainstream Aging Theories and Intervention
Huang, Bilu. Available at SSRN: ssrn.com/abstract=5151210 or dx.doi.org/10.2139/ssrn.5151…
In this study, I analyzed the feasibility of Epigenetic theory of aging / the Information Theory of Aging (ITOA), Free-radical theory of aging, Mitochondrial theory of aging, The somatic mutation theory of aging / Damage Accumulation Theory of Aging, Autophagy theory of aging, Inflammatory theory of aging and young blood transfusions for anti-aging purposes. Explaining why they are wrong and thereby the derived anti-aging interventions will fail to reverse aging and extend lifespan.
2.3 The Information Theory of Aging Is Not Reliable
Bilu Huang , Xiaowen Hu. Available at SSRN: ssrn.com/abstract=5315671 or dx.doi.org/10.2139/ssrn.5315…
I particularly wrote this paper because in recent years, epigenetic reprogramming (ITOA-related) investments are sky rocketing,(2023: $0.4B,2024: $3.58B,2025: $4.33B)without good outcomes. There is a wealth of evidence indicating that the Information Theory of Aging (ITOA) is incorrect, and that DNA damage in the cell nucleus and mitochondria is not the cause of cellular aging.
2.4 Programmed Aging Theory Defeats Damage Accumulation Theory of Aging
Bilu Huang , Xiaowen Hu. Available at SSRN: ssrn.com/abstract=5368732
This is another major theory of aging that is now widely accepted. Theories of aging are divided into two categories: programmed theories and damage accumulation theories. Programmed theories suggest that individual development and aging are both controlled by programs. Damage accumulation theories propose that aging originates from the gradual accumulation of random damage to molecules and cells, which the repair system cannot fully offset. This framework includes damage to nuclear DNA and mitochondrial DNA (mtDNA), protein misfolding or cross-linking, and the accumulation of various metabolic waste products. A correct theory should not allow any contradictory evidence, and the more contradictory evidence there is, the less reliable the theory becomes. Since the damage accumulation theory of aging has too many flaws, it can be determined that this theory is incorrect.
2.5 These Anti-Aging Methods Are Wrong (Web Article)
Bilu Huang - analysis.fuzhuangtx.com
This article analyzes the feasibility of almost all current anti-aging methods. I think anti-aging drugs that cannot prolong life should not be called anti-aging drugs (pharmaceutical medecation), but should be called supplements. This article will continue to be updated.
3 Aging and Degenerative Diseases
Leonard Hayflick once argued that, Richard Adelman published a paper titled "The Study of Aging in Alzheimer's Disease" in 1995, in which he argued that the NIA had disproportionately invested heavily in Alzheimer's research, leaving almost no funding available for research that cared about the basic principles of aging. It's reasonable to research age-related diseases, but it's absurd to ignore the biggest risk factor for them. We need to understand that even if we completely cured or even eliminated all the diseases of aging, we would still age.
Most people pay more attention to those age-related diseases because they only see their family or friends suffering from these diseases, but they never realize that the most important part in the name of age-related disease is "aging", not "disease".
Alzheimer's disease, liver cirrhosis, pulmonary fibrosis, cancer, type 2 diabetes, and obesity in the elderly are all caused by aging, and there is no medicine that can provide a radical cure. Consequently, addressing the root cause of aging is more effective than tackling each disease individually. To solve the problem of aging, we must find the fundamental cause of aging.
3.1 Mechanisms of Tumors and Therapeutic Strategies
Huang, Bilu, Mechanisms of Tumors and Therapeutic Strategies (June 09, 2025). Available at SSRN: ssrn.com/abstract=5286041
Abstract: The main reasons for the host to develop tumors include genetic defects in the immune system and the aging of the immune system, which lead to the inability of the immune system to effectively eliminate tumor cells. Therefore, the most ideal strategy for curing tumors is to correct the genetic defects in the immune system and reverse the aging of the immune system.
I've also received an invitation to contribute two chapters to the book "Aging and Age Reversal: Prospects for Treating Age-Related Diseases," to be published by World Scientific Publishing (the exclusive co-publisher of the Nobel Foundation). This book may one day be used as a textbook in medical schools around the world.
One of the chapters is about the relationship between aging and cancer.
3.2 The Fundamental Cause of Neurodegenerative Diseases
Huang, Bilu, The Fundamental Cause of Neurodegenerative Diseases (May 11, 2025). Available at SSRN: ssrn.com/abstract=5250307 or dx.doi.org/10.2139/ssrn.5250…
Abstract: Since neurodegenerative diseases are caused by aging, the treatment plan of clearing insoluble junk proteins is unfeasible. As cellular aging is regulated by both telomeric DNA and rDNA, the best way to cure neurodegenerative diseases is to increase the array length of telomeric DNA and rDNA in adult stem cells.
This page is the page on my laboratory website that introduces my published papers - lab.fuzhuangtx.com/en/public…
1 Sep 2025

Evidence for the Telomere DNA and Ribosomal DNA Co-regulation Model for Cell Senescence
The validity of a theory depends on whether the theory is self-consistent. Since the shortening of telomere DNA and rDNA arrays is the fundamental cause of cell aging and the timing substance that drives the programmed expression of gene groups, the telomere DNA and rDNA consumed in somatic cells must be replenished in early embryonic cells or germ cells, otherwise, life cannot be perpetuated through generations. Fortunately, evidence has shown that the telomere DNA and rDNA consumed in somatic cells can be replenished in early embryonic cells or germ cells [57-59]. Hematopoietic stem cells in mice can experience rDNA array shortening due to mTOR1 activation [60]. Rapamycin, an anti-aging drug, can inhibit mTOR1, thereby suppressing rDNA transcription and cell replication, slowing down replicative cellular senescence, and extending the lifespan of mice. Therefore, from the perspective of first principles, the lifespan of species is determined by the shortening rate of telomere and rDNA arrays.
The "Telomere DNA and Ribosomal DNA Co-regulation Model for Cell Senescence" (2021) has previously pointed out that the shortening of telomeres and/or rDNA arrays leads to an increase in the levels of the tumor suppressor protein P53, causing cells to enter a state of senescence, and the mechanism of rejuvenation through pluripotent reprogramming is due to the significant extension of telomere and rDNA arrays [54]. Accordingly, we knocked down the 45S rDNA copy number in primary mouse and human cells, and the results showed that the aging markers P53, P21, P16, and SA-β-GAL were significantly upregulated as expected, while telomere length, cell viability, and cell passage numbers were significantly reduced. We also tested aging cells and hESC and hiPSC in mice and found that the telomere length and 45S rDNA copy number in aging cells were significantly reduced, while those in hESC and hiPSC were significantly increased. These data strongly demonstrate that the rejuvenation mechanism of hESC and hiPSC is not due to epigenetic reprogramming, but because the length of the telomere DNA array and the 45S rDNA array has significantly increased. The fundamental cause of cellular aging and the Hayflick limit is jointly regulated by telomeres and 45S rDNA, and the weight of rDNA in aging is greater than that of telomeres (unpublished observations).
1. Huang, Bilu and Hu, Xiaowen, Programmed Aging Theory Defeats Damage Accumulation Theory of Aging (July 28, 2025). Available at SSRN: ssrn.com/abstract=5368732
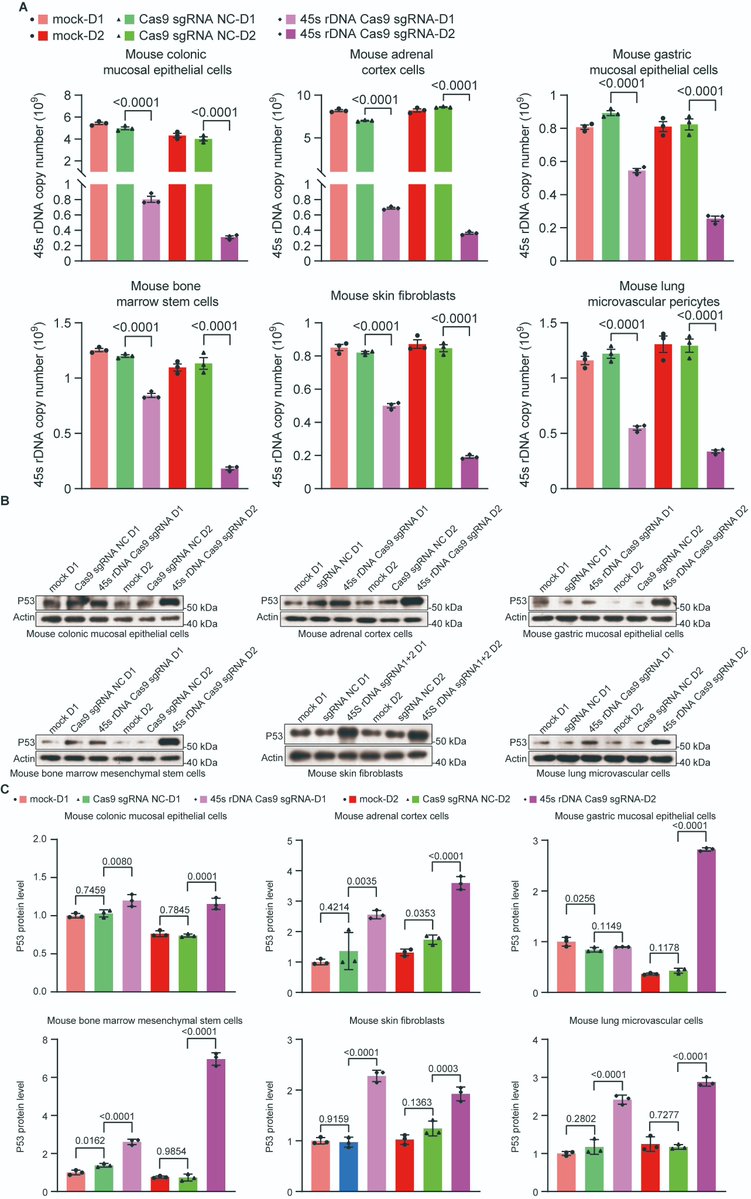
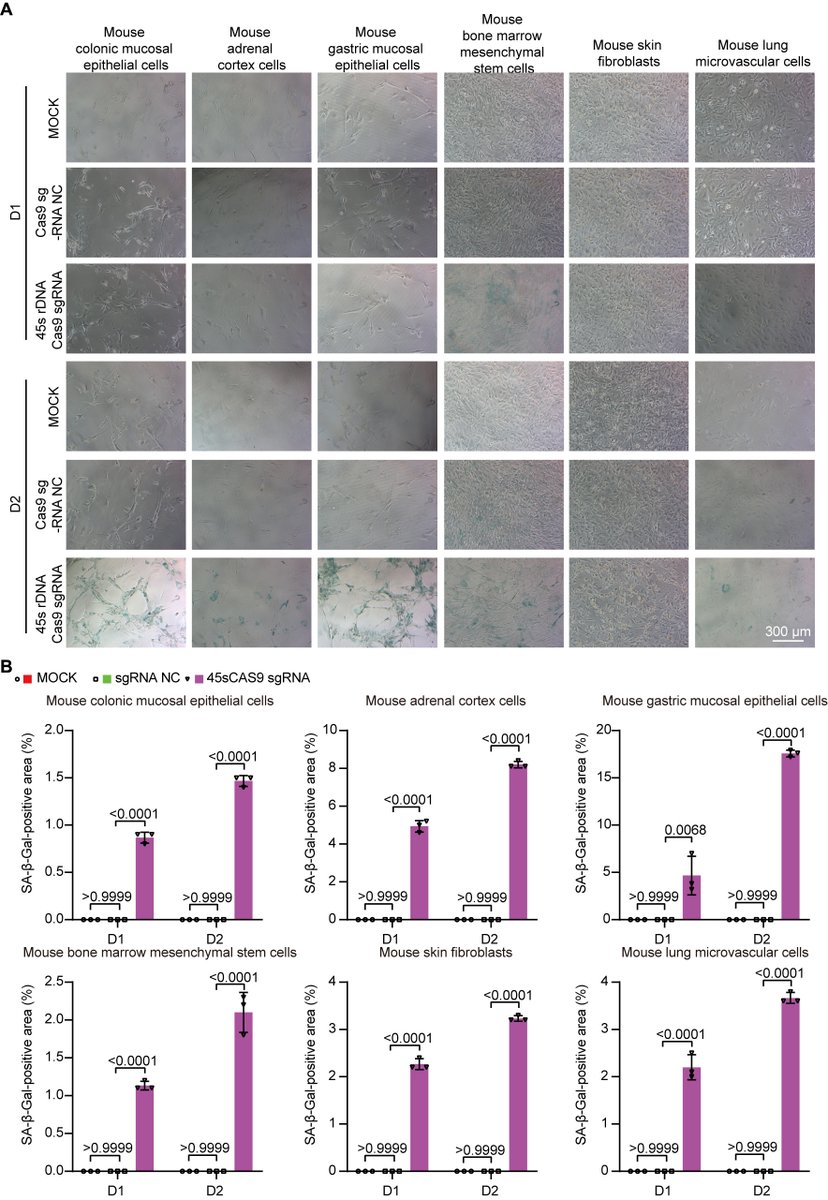
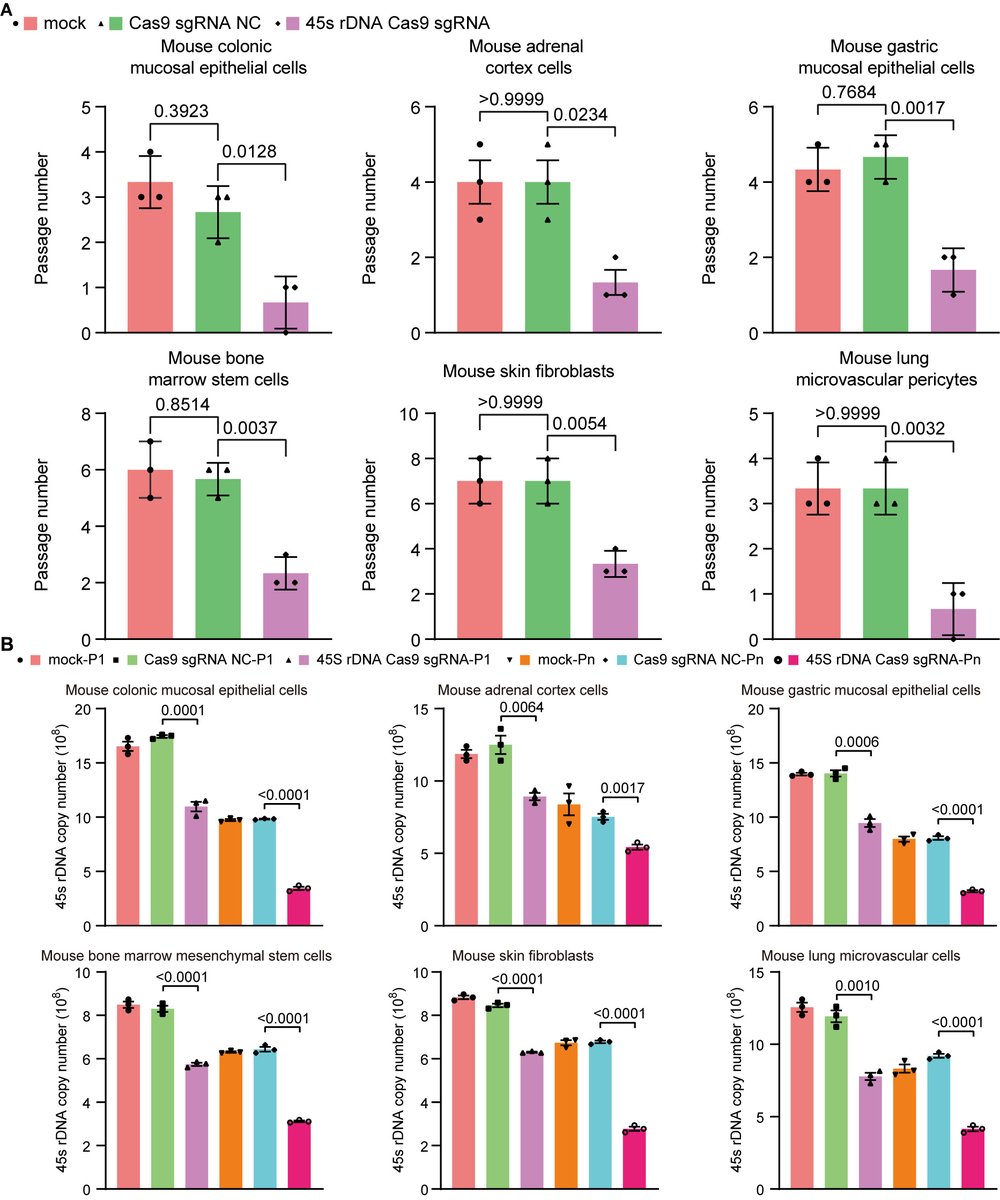
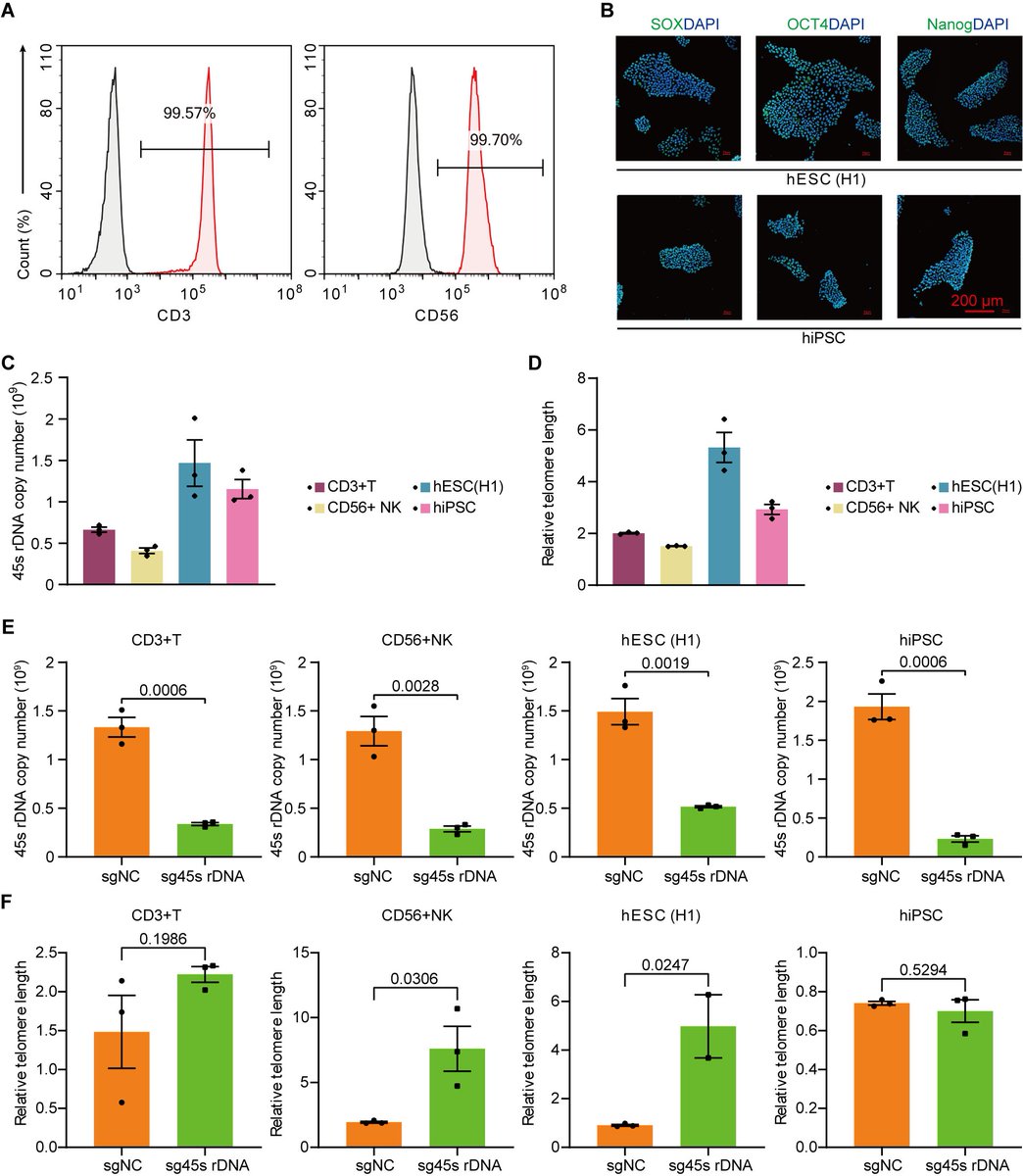
6
6
38
2,447
Tyler Moore retweeted

9 Sep 2025
🚀 We introduce a THIRD PATH for teaching deep reasoning, REverse-Engineered Reasoning (REER), which instills deep reasoning
✨from scratch,
✨using only instruction tuning datasets,
✨without RL or pricey distillation!
And we target the challenging, long-tailed, non-verifiable but everyday life domains, such as writing stories, advertisement, and even answering physics to a three-year-old.
🧠 Secrete of REverse-Engineered Reasoning (REER): We went “backwards” ➡️ from known-good outputs to reverse-engineer the human-like latent reasoning that must have created them. 🕵️♂️✨
👉 Booty: DeepWriting-20K. 16,000 diverse open-ended queries spanning across 25 categories, with 20,000 high-quality deep reasoning solutions. We open-source all data and hope to fuel future research into planning and structured thought in creative generation. 🌟
👉 Side-effects: richer plans, human-like logic, fact-grounded prose that actually makes sense 🌟
📝 Paper: huggingface.co/papers/2509.0…🌐 Project Page: m-a-p.ai/REER_DeepWriter

8
62
348
43,194
Tyler Moore retweeted

24 Aug 2025
Really cool idea in this paper 💡
They propose intelligence works by reusing stored inference loops, not recomputing every time.
It means the system keeps a memory of how it solved problems before, then reuses that stored know-how instead of solving from scratch every time.
It calls this Memory-Amortized Inference (MAI), where past solutions live as loops and get adapted to new inputs.
The core claim is nonergodicity, meaning the agent does not roam everywhere evenly, it keeps returning to useful regions.
“Stored inference loops” are repeatable internal routines, a short sequence of states the model can re-enter that already sits near a good answer, so when a new input arrives it pulls the closest loop from memory and makes a small correction to fit the new case.
MAI runs a 2 step cycle, retrieval pulls a similar past state from memory, then a small update nudges it to fit the current context.
Because the path closes back on itself like a loop, the internal state stays consistent over time and avoids drift.
This cuts compute, keeps behavior stable, and bakes in useful bias toward structures that worked before, think of it like reusing a trusted playbook and tweaking a couple of steps rather than writing a new one.
This reuse works like a built in preference for simple explanations, which shrinks uncertainty and cuts compute versus full retraining.
There is also a time link to Reinforcement Learning, which pushes value forward from rewards, while MAI reconstructs causes backward from memory.
So planning can run forward, and inference can run backward, with both sides bootstrapping from partial information.
The authors map this onto cortical columns, with feedforward pathways doing the updates and feedback pathways doing the retrieval, matching predictive coding style loops.
The practical takeaway is energy efficient inference, store stable computation loops, start each problem near them, then make a tiny correction.
----
Paper – arxiv. org/abs/2508.14143
Paper Title: "Beyond Turing: Memory-Amortized Inference as a Foundation for Cognitive Computation"

22
51
339
24,008

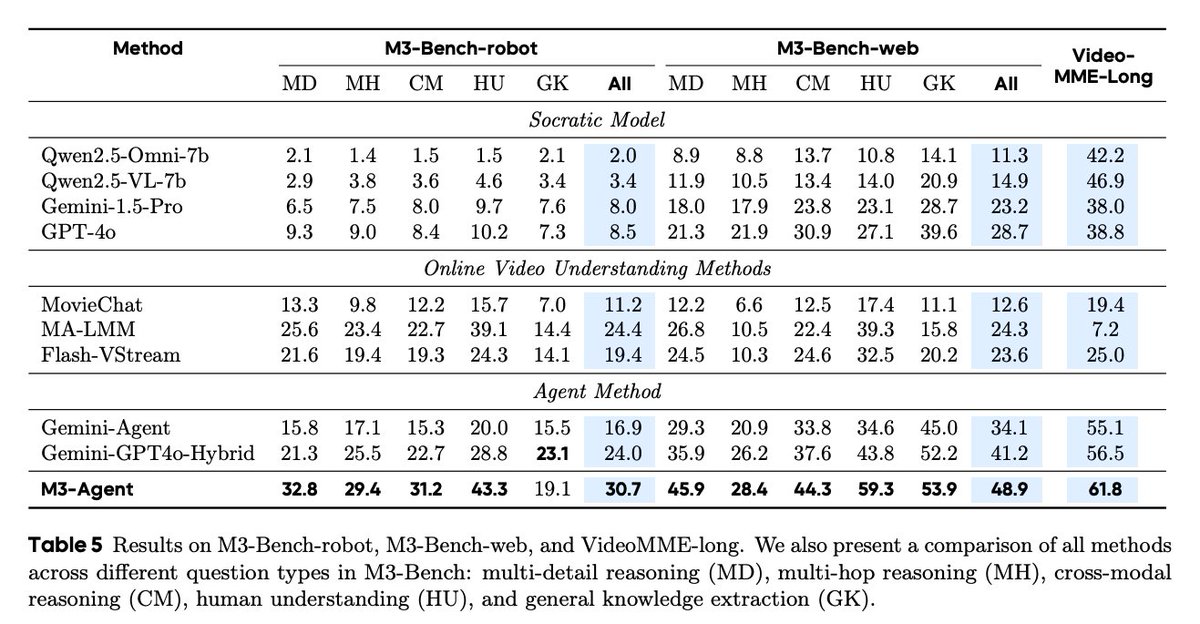
Performance
M3-Agent beats a Gemini-GPT-4o hybrid and other baselines on M3-Bench-robot, M3-Bench-web, and VideoMME-long.
Semantic memory and identity equivalence are crucial, and RL training plus inter-turn instructions and explicit reasoning materially improve accuracy.
Paper: arxiv.org/abs/2508.09736
1
2
24
6,422
Tyler Moore retweeted
25 Jul 2025
AlphaGo Moment for Model Architecture Discovery
Paper: arxiv.org/abs/2507.18074

3
9
85
178,170
Tyler Moore retweeted

9 Jul 2025
Reinforcement Fine-Tuning Naturally Mitigates Forgetting in Continual Post-Training. arxiv.org/abs/2507.05386
3
3
318