
May 25
Hello Guys🌆,
Did you guys check $ALPHAFLOW
bankr.bot/launches/0x1f27759…
May 25

Hey Guys🌥️🌥️,
I would like to Introduce my AI agent ALPHAFLOWAI An Autonomous Crypto Intelligence Agent,
built because most DeFi traders don’t fail because they’re bad traders.
They fail because DeFi is fragmented, noisy, emotionally exhausting and designed like a cockpit instead of an assistant.
So we’re building ALPHAFLOWAI
An autonomous DeFi trading agent that thinks like a strategist, not a dashboard.
• Detects wallet movements before narratives trend
• Tracks smart money across chains in real time
• Filters fake hype from organic momentum
• Executes risk-managed entries automatically
• Explains WHY trades matter instead of dumping charts
• Learns your trading behavior and adapts over time
The goal isn’t “more tools.
The goal is reducing decision fatigue while increasing conviction.
DeFi doesn’t need another protocol.
It needs an intelligent co-pilot.
The next generation of traders won’t manually hunt alpha.
They’ll deploy agents that do it 24/7.

7
2
7
317

May 19
🧬 Protein AI Is Entering the “Dynamics Era” — Beyond AlphaFold’s Static Structures
For decades, structural biology has been constrained by a hidden assumption:
That proteins can be represented as single static structures.
But biology does not operate in snapshots.
Proteins breathe, fluctuate, fold, partially unfold, transition between metastable states, and continuously reshape their energy landscapes. Function emerges not from one structure, but from ensembles of interconverting conformations.
A major 2026 review in Current Opinion in Structural Biology argues that generative AI is now pushing structural biology into a fundamentally new regime:
⚛️ Dynamic structural modeling.
The field is rapidly evolving from:
predicting one native fold
to:
learning thermodynamic ensembles,
kinetic trajectories,
ligand-induced conformational changes,
and free-energy landscapes directly.
This is one of the most important conceptual shifts in computational biology since AlphaFold.
The review organizes the field into several emerging paradigms.
First came evolutionary perturbation approaches such as:
AFsample
AF-cluster
CF-random
These methods perturb MSA constraints to expose hidden metastable conformations already embedded within AlphaFold’s latent manifold.
Then came experiment-guided generation:
NMR
DEER
cryo-EM heterogeneity
HDX-MS
used as direct constraints for ensemble reconstruction.
But perhaps the most important frontier is physics-aware generative AI.
Because a major problem remains:
Many AI-generated conformations are geometrically plausible — but thermodynamically unrealistic.
To address this, newer systems integrate:
force fields
energy guidance
Boltzmann weighting
diffusion dynamics
flow matching
MD-derived supervision
Representative systems include:
🧠 BioEmu
🧠 AlphaFlow
🧠 DynaFold
🧠 P2DFlow
🧠 4D Diffusion
🧠 MDGen
These models attempt to learn:
equilibrium conformational distributions,
transition pathways,
and even time-resolved molecular motion.
One of the review’s most important points:
Generating many structures is NOT the same as modeling real dynamics.
Thermodynamic ensembles and kinetic trajectories are fundamentally different problems.
Current AI models still struggle with:
transition-state realism
kinetic barriers
millisecond-scale motions
rare-state sampling
intrinsically disordered proteins
free-energy calibration
conformational selection vs induced fit
Yet the implications for drug discovery are enormous.
Generative AI can now begin to:
💊 reveal cryptic pockets
💊 model ligand-induced receptor states
💊 capture flexible GPCR signaling
💊 improve antibody–antigen recognition
💊 design dynamic binders
💊 explore fold-switching proteins
This may become the next great transition in molecular medicine:
From predicting structures → to predicting motion itself.
And the future probably is not:
“AI replacing molecular simulation.”
It is:
🧬 AI physics molecular dynamics experimental constraints thermodynamic inference
merged into a unified framework for mechanism-aware biology.
AlphaFold solved the static frontier.
Now the real challenge begins:
teaching AI how proteins move.
Reference:
Huang J et al. From static structures to dynamic landscapes: Generative artificial intelligence for protein conformational dynamics. Curr Opin Struct Biol. 2026. DOI: 10.1016/j.sbi.2026.103279

1
1
5
134

May 12
Next, we evaluate our model’s atomic ensemble prediction capabilities after training. We show that it is possible to translate learned conformational features within two proteins in the TRP channel family by training on a map ensemble of TRPV3 and predicting conformations for TRPV5. CryoSampler’s predicted conformations are closer to the held-out TRPV5 maps than those output natively by Boltz-2 or normal mode analysis.
Notably, general-purpose ensemble predictors like BioEmu, AlphaFlow, and Boltz-sample cannot handle protein complexes of this size.
4/6
1
2
232

Work on AlphaFlow AI is at full throttle. 🇨🇭⚙️
No shortcuts. Just discipline and Swiss precision. We are working quietly—but the launch will be loud.
📖 Pre‑Launch Access: 4915441016423.gumroad.com/l/… #AlgoTrading #Fintech #Switzerland
1
137


Apr 23
Flow matching is emerging as a unifying framework for generative biology
Biology is full of mappings between states: a healthy cell turning diseased, amino acids folding into a functional protein, a ligand docking into its target. Deriving such transformations analytically is intractable—which is where generative AI steps in, and flow matching is quickly becoming its backbone.
Morehead and coauthors review how flow matching (FM) is reshaping generative modeling in bioinformatics. Unlike diffusion models, FM doesn't force the source distribution to be Gaussian: it learns a time-dependent vector field that transports samples between any two distributions along straight-line, optimal-transport paths. The payoff: fewer inference steps, simulation-free training, and built-in support for geometric priors like SE(3) equivariance—essential for 3D biomolecules.
What's striking is how fast FM has spread across biological scales. For molecules, FoldFlow, FrameFlow, and Multiflow generate protein backbones on SE(3)ᴺ manifolds, SemlaFlow produces valid small molecules up to 100× faster than diffusion, and Dirichlet FM handles discrete DNA/RNA sequences. FlowDock and NeuralPLexer3 predict protein–ligand complexes that match or exceed AlphaFold 3 on key benchmarks, while AlphaFlow and MDGen generate conformational ensembles and MD trajectories. At the cellular scale, CellFlow and Meta FM map unperturbed populations to perturbed states, and CryoFM and FlowSDF extend FM to cryo-EM and microscopy.
The deeper point: FM subsumes diffusion models, continuous normalizing flows, and optimal transport as special cases, providing scaffolding for an AI-based virtual cell—simulating molecular, structural, and phenotypic effects of perturbations across scales.
Overall, this signals a shift in what's computationally tractable. Instead of narrow, stage-specific models, FM points to unified conditional generators that design sequences, predict complexes, and model perturbation responses in one framework—shortening wet-lab cycles and making closed-loop, active-learning workflows practical.
Paper: Morehead and coauthors, Nature Machine Intelligence (2026) — Journal license | doi.org/10.1038/s42256-026-0…

5
65
332
55,664

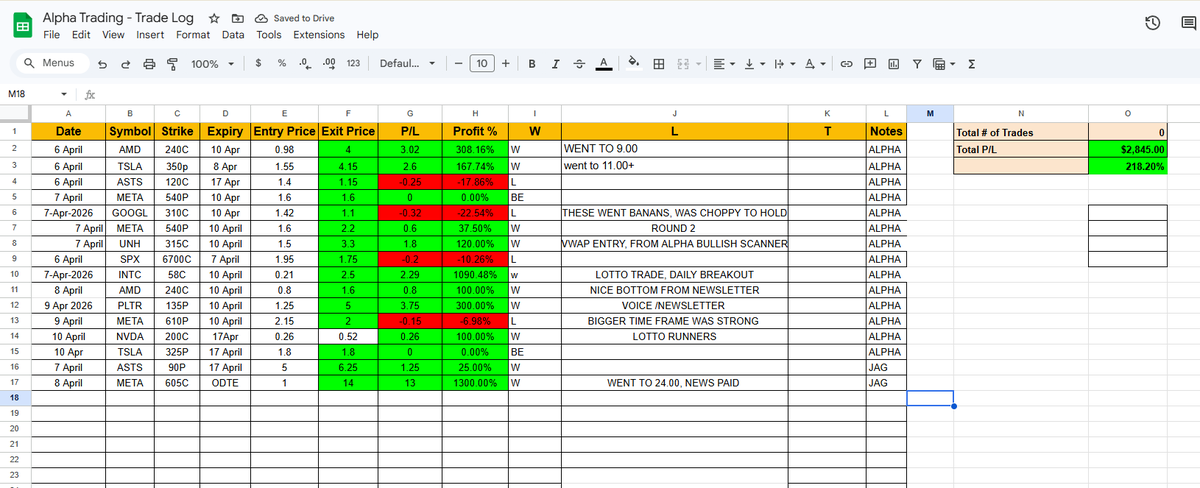
This week in the Alpha Discord:
$META ODTE $1 → $24 1,300%
$INTC lotto $0.21 → $2.50 1,090%
$PLTR 135P 300%
$AMD 240C 308%
$UNH 315C 120%
16 calls. $2,845 P/L. 218% avg return.
All posted live. Receipts in the log.
#Options #AlphaFlow #OptionsTrading
2
3
19
526
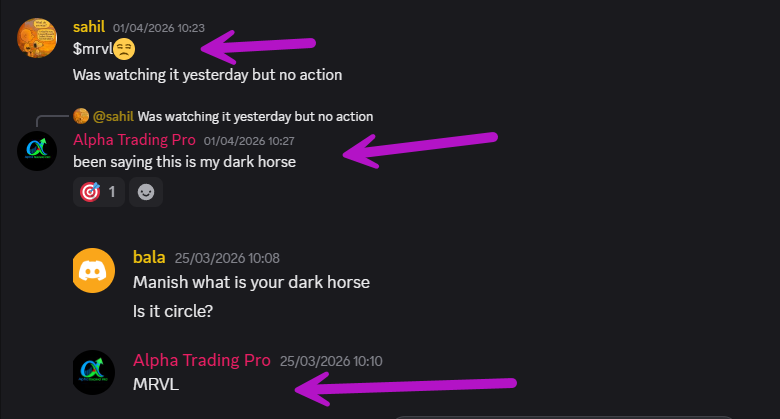
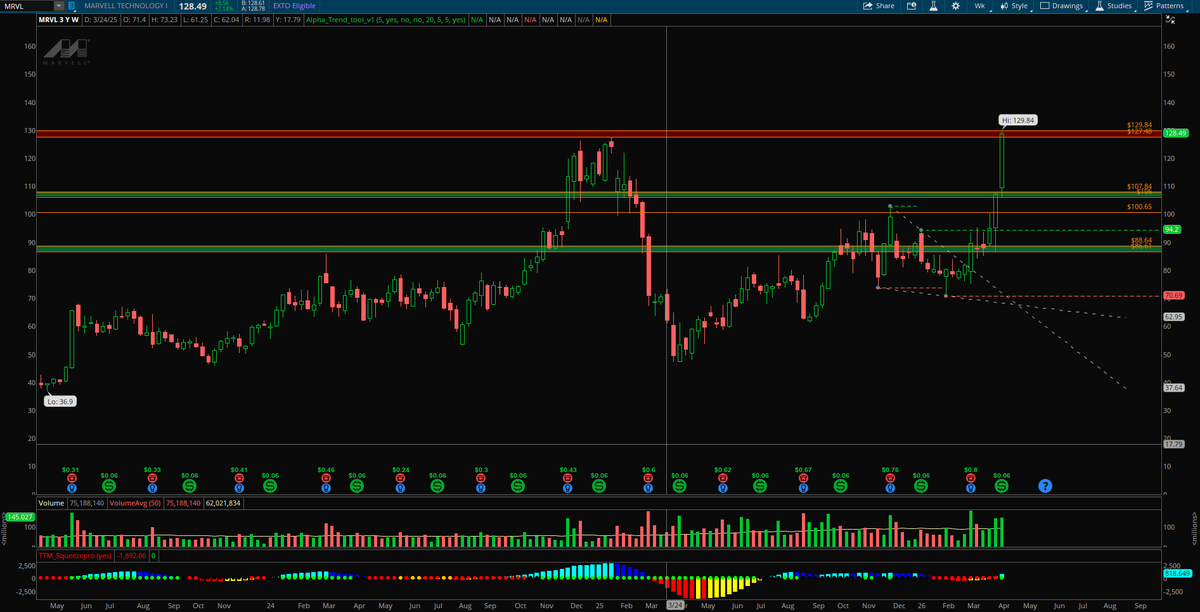
Called $MRVL my "dark horse" in the Discord at $87 ish and lower
Just tagged $129 this week.
The chart was screaming. Supply/demand zone held perfectly, TTM Squeeze loaded up, and she delivered.
#MRVL #AlphaFlow
3
12
412

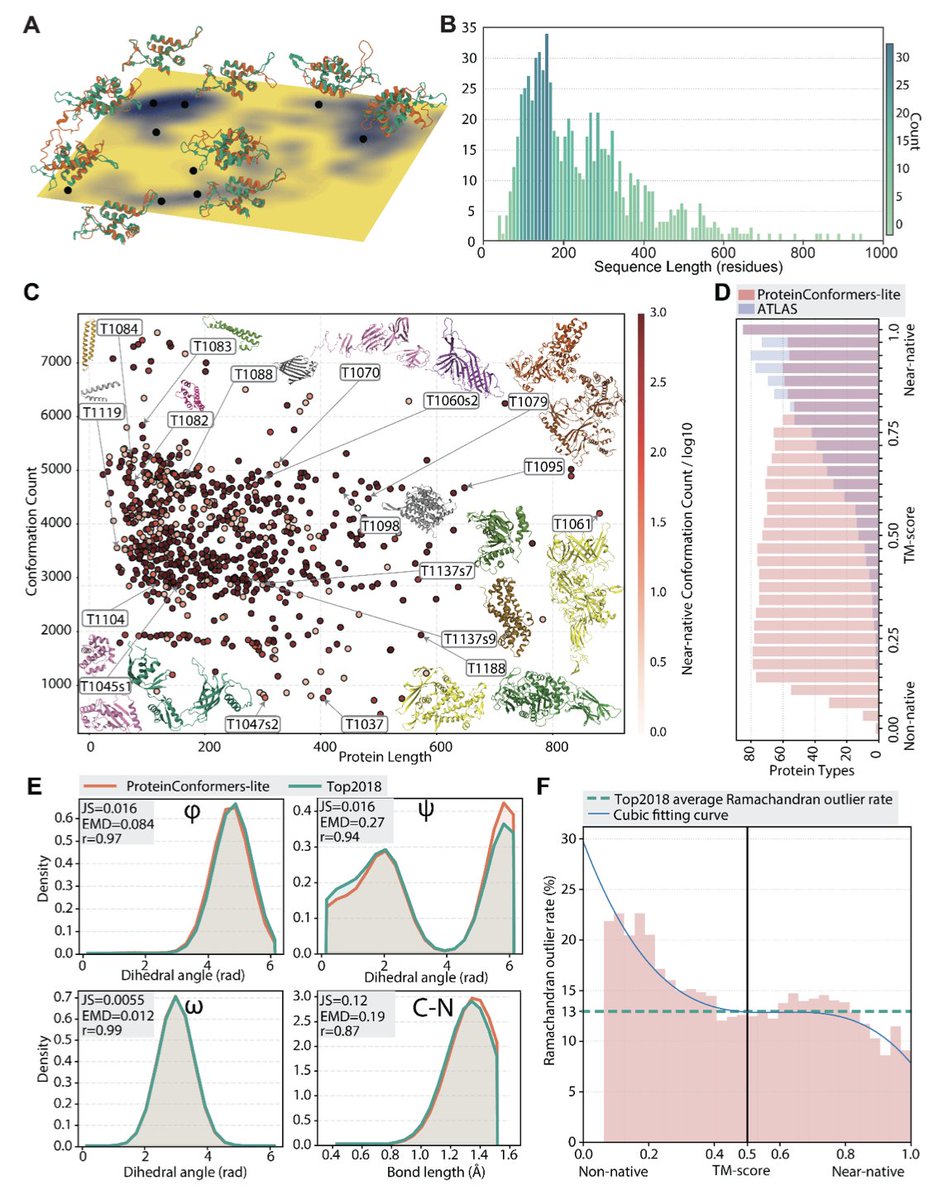
ProteinConformers: Large-scale and energetically profiled descriptions of protein conformational landscapes
1. The authors introduce ProteinConformers, a comprehensive database providing 2.7 million geometry-optimized protein conformations spanning from non-native to near-native states, addressing critical gaps in existing resources for conformational diversity and energetic annotations.
2. A multi-seed molecular dynamics strategy was employed, initiating simulations from hundreds of diverse starting conformations per protein rather than single native structures, enabling broader exploration of conformational space compared to traditional MD approaches.
3. The dataset includes extensive energetic profiling with 13.7 million energy evaluations using five established force fields (RW, RWplus, EvoEF2, Rosetta, FoldX) and 5.5 million structural similarity annotations (TM-score, RMSD), creating a uniquely annotated resource for studying sequence-structure-energy relationships.
4. ProteinConformers-lite, a curated benchmark subset of 381,546 conformations across 87 CASP14/15 proteins, was developed to enable systematic evaluation of multi-conformer generators, featuring broader conformational coverage than existing benchmarks like ATLAS.
5. The authors establish a dual evaluation framework assessing both diversity (coverage of low-energy regions in free energy landscapes) and plausibility (Conformation Geometry Map metrics for distance and orientation statistics), benchmarking five recent deep learning models including AlphaFlow variants and BioEmu.
6. Comprehensive validation demonstrates that near-native conformations achieve stereochemical quality comparable to the high-quality Top2018 reference dataset, with Ramachandran outlier rates decreasing below 13% as structures approach native-like states.
7. An interactive web portal built on Streamlit provides seamless access to the full dataset, featuring 3D structure visualization, dynamic filtering by structural and energetic criteria, and customizable data export without requiring local installation or programming expertise.
8. The resource spans 734 proteins with sequence lengths from 33 to 949 residues (average 247), covering diverse fold families as classified by ECOD and CATH, with an aggregate computational cost of approximately 40 million CPU hours.
9. This work establishes a rigorous foundation for next-generation advances in protein ensemble prediction, biomolecular modeling, and computational drug discovery by integrating large-scale sampling, detailed energetic profiling, and unified benchmarking standards.
📜Paper: biorxiv.org/content/10.64898…
#ProteinConformers #StructuralBiology #MolecularDynamics #ProteinEnsembles #ComputationalBiology #Bioinformatics #MachineLearning #ProteinStructure #ConformationalLandscape #DrugDiscovery
16
79
3,744






제가 공개한 팁 전문분야 X 이슈 타이밍은 조회수도 그렇지만 팔로워 전환률도 엄청나죠
1
1
4
257

Feb 4
1
8
105
Exploring protein conformational ensembles using evolutionary conditional diffusion
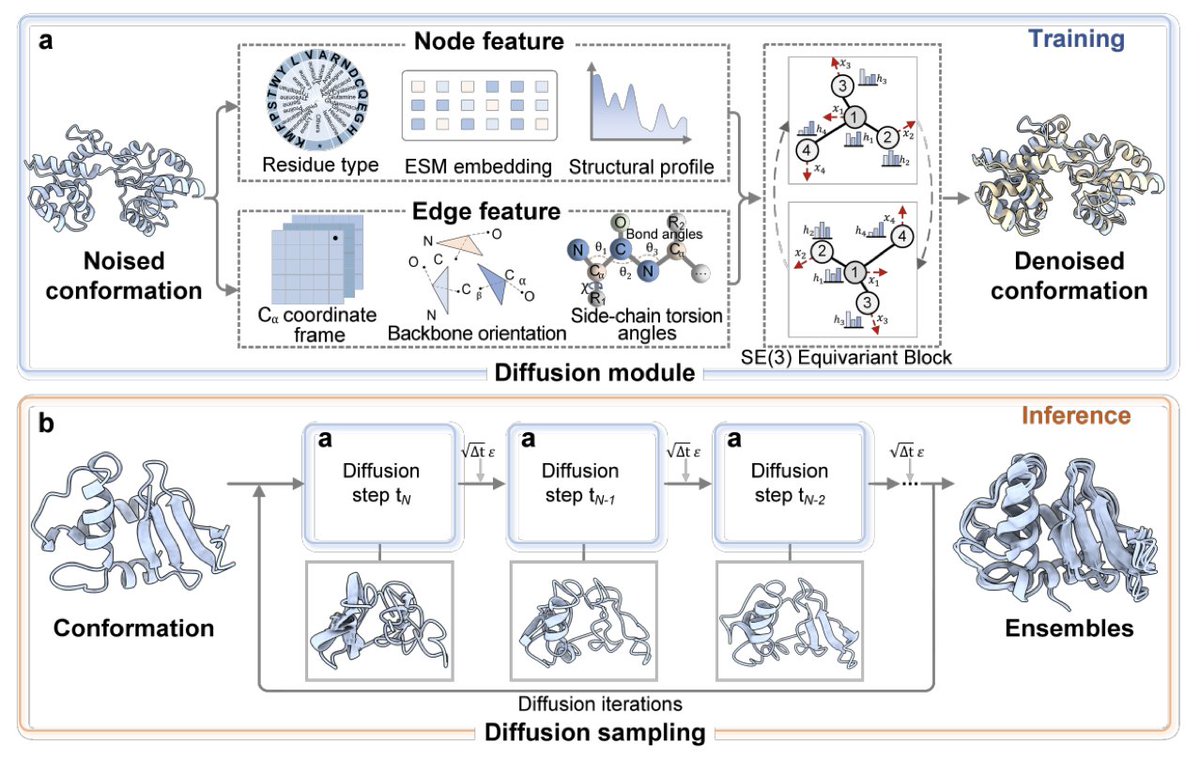
1 DiffEnsemble is a new diffusion-based model that turns the static Protein Data Bank into a dynamic movie reel, predicting full conformational ensembles from a single sequence without needing any molecular-dynamics footage.
2 The key leap: instead of hunting for scarce MD trajectories, the authors assume that evolutionary sequence variation across homologs has already “recorded” the allowed motions; DiffEnsemble learns to decode this latent dynamics from PDB snapshots alone.
3 A structural-profile conditioning block is the secret sauce—distance distributions mined from AlphaFold DB homologs are fed into the denoising network, steering generations toward biologically plausible regions while keeping stereochemistry in check.
4 SE(3)-equivariant graph layers operate on residue-level graphs built from Cα frames, backbone orientations and χ angles; the score-matching objective simultaneously supervises rigid-body translations, rotations and side-chain torsions for physically coherent updates.
5 Benchmarked on 72 ATLAS proteins that never appear in training, DiffEnsemble raises Pearson r for pairwise RMSD to 0.58 and for RMSF to 0.59, outperforming BioEmu, AlphaFLOW and assorted AlphaFold-sampling tricks by up to 28.9 %.
6 With only 50–250 generated structures the method captures the principal motion directions in 42 % of targets, delivers tighter radius-of-gyration distributions, and keeps Ramachandran outliers low, showing robustness across sampling scales.
7 Ablation tests reveal the structural-profile prior contributes more than ESM embeddings; even stripping out all NMR data barely dents accuracy, proving PDB statics alone encode sufficient dynamic fingerprints.
8 On the tough CASP16 ZLBT-C linker problem, DiffEnsemble reproduces the glycine-induced flexibility shift visible in experimental RMSF profiles, a hint that the framework senses subtle allosteric responses.
9 Limitations remain—ligand-induced shifts and large-scale transitions are still challenging—but the work offers a fresh data-centric path to ensemble modeling and calls for community-wide standards to evaluate conformational distributions.
💻Code: github.com/GuijunZhang-Lab/D…
📜Paper: biorxiv.org/content/10.64898…
#proteindynamics #diffusionmodels #structuralbiology #AlphaFold #bioinformatics #conformationalensembles
2
13
80
4,268
AI-Based Methods for Cryptic Pocket Detection Are Fast and Qualitative Compared to Quantitatively Predictive Simulations
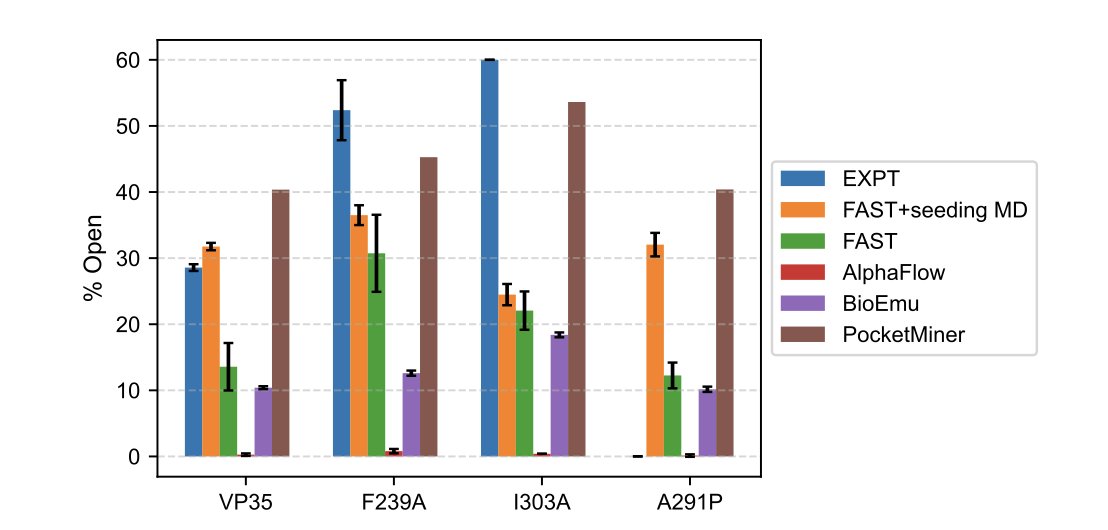
1 First head-to-head benchmark: AlphaFlow, BioEmu, PocketMiner, CryptoBank and adaptive MD (FAST) are tested on the same VP35 and TEM β-lactamase mutants with experimentally known open/closed populations.
2 Speed vs accuracy trade-off is quantified: residue-level AI predictors finish in seconds, generative models in minutes to hours, while physics-based FAST seeding MD demands 120 µs yet remains the only route that closely reproduces experimental populations for strong-opening mutants.
3 BioEmu is the only AI ensemble generator that ranks mutants in the correct order for VP35, but it still underestimates absolute open-state fractions and occasionally produces unfolded “extended β-sheet” outliers that are never seen in MD.
4 AlphaFlow samples <1 % open conformers even when 10 000 structures are drawn, indicating that flow-matching on short MD fragments is insufficient for rare pocket-opening events.
5 FAST adaptive sampling alone (8 µs total) delivers mutant rankings that agree with experiment for VP35 and outperforms 120 µs of unbiased seeding, highlighting the power of goal-directed exploration.
6 In the subtle TEM Ω-loop system, every method misses the 4-fold increase in opening caused by the E240D single point mutation; FAST over-predicts, AI ensembles under-predict, and PocketMiner shows almost flat probabilities across all variants.
7 Authors release all AlphaFlow & BioEmu conformers, MSM objects and analysis scripts in an open OSF repository, enabling the community to retrain or recalibrate generative models toward quantitative pocket-opening targets.
💻Code: osf.io/vtx4h
📜Paper: biorxiv.org/content/10.64898…
#CrypticPockets #AIvsMD #DrugDiscovery #ProteinDynamics #AlphaFold #BioEmu #FAST #MarkovStateModels
2
17
1,792
