CAPTAIN: A multimodal foundation model pretrained on co-assayed single-cell RNA and protein
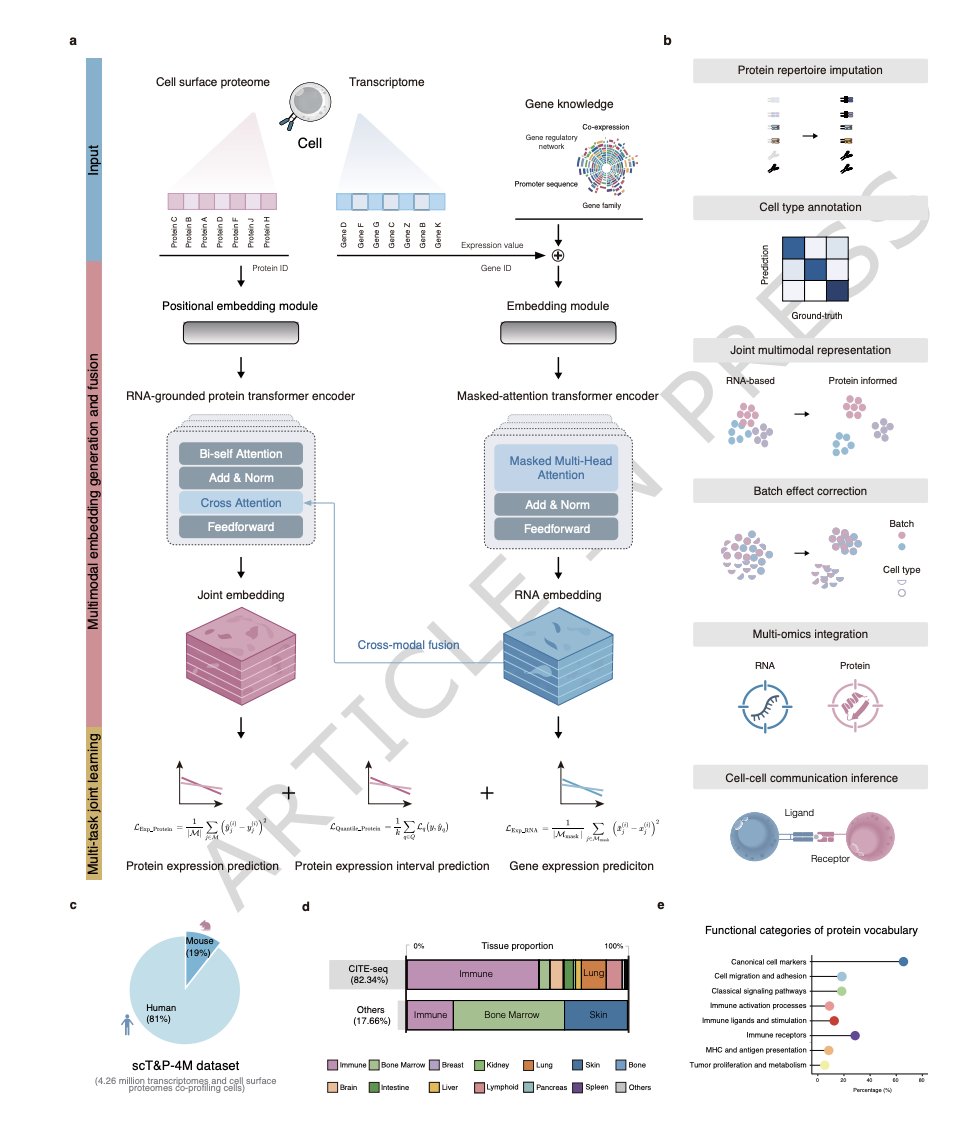
1. The paper introduces CAPTAIN, a multimodal single-cell foundation model that learns unified cell representations from co-assayed RNA surface protein data, aiming to reduce the bias of transcriptome-only foundation models when protein phenotypes drive cell state.
2. A key enabling contribution is the scT&P-4M pretraining corpus: 4.26 million paired single cells from human and mouse, spanning 13 tissues, 249 samples, multiple platforms (mostly CITE-seq), and a harmonized vocabulary of 382 curated surface proteins with standardized naming and functional categorization.
3. Model design: a dual-encoder Transformer (RNA encoder protein encoder) with cross-modal attention to align modalities into a shared latent space. The RNA encoder is initialized from scGPT (pretrained on 33M scRNA cells) and augmented with a gene-knowledge module encoding priors (GRN, promoter sequence, gene family, co-expression), plus species tokens for human/mouse.
4. Pretraining objective is explicitly multimodal and multi-task: masked gene expression reconstruction (unsupervised), protein abundance prediction (supervised over measured proteins), and protein prediction interval/uncertainty estimation via quantile regression—so proteins are treated as first-class signals, not just auxiliary outputs.
5. Protein inference is framed in two settings: fine-tuned prediction (adapt on a subset of paired cells from a new dataset) and zero-shot prediction (no dataset-specific paired training). Predictions are restricted to the fixed 382-protein vocabulary (not open-vocabulary), but can include proteins absent from a study’s antibody panel.
6. On protein imputation/expansion benchmarks across diverse datasets (e.g., human PBMC, MALT, monocytes; mouse PBMC), CAPTAIN reports consistently strong performance vs Seurat, sciPENN, TotalVI, scTranslator, and scTEL, with competitive zero-shot behavior and broader coverage when other methods cannot produce predictions for many proteins.
7. For cell type annotation, CAPTAIN fine-tuning adds a classifier on learned embeddings (RNA-only or multimodal). It reports 96.1% accuracy on PBMC CITE-seq, and shows particular strength in fine-grained T cell subtype labeling in bone marrow (Macro-F1 0.73 vs Seurat 0.61 and scGPT 0.04), emphasizing the value of protein-aware representations when RNA alone is insufficient.
8. For integration/batch harmonization, CAPTAIN is evaluated on multi-batch scRNA-seq and multi-omic datasets, aiming to balance batch removal with biological conservation. It is also tested on difficult cross-platform settings, aligning >60,000 cells across CITE-seq, ECCITE-seq, and TEA-seq, with additional metrics highlighting the remaining difficulty of full cross-technology mixing.
9. A notable downstream application is protein-informed cell–cell communication: CAPTAIN imputes receptor abundance (protein) while using ligand expression from RNA, then performs ligand–receptor inference with permutation testing. In PBMCs, it prioritizes 22 interactions from CD4 naive T to NK cells, with 18/22 (81.8%) supported by literature, and validates predicted signaling by showing upregulation of NicheNet-prioritized target genes in “strong communication” receiver cells.
10. In a COVID-19 multi-sample analysis, CAPTAIN suggests severity-associated increases in platelet-to-monocyte signaling and highlights a protein-driven S100A9–CD36 axis that may be missed by transcriptome-only approaches due to post-transcriptional decoupling; the paper further assesses structural plausibility with protein–protein docking.
📜Paper: doi.org/10.1038/s41467-026-7…
#SingleCell #CITEseq #MultiOmics #FoundationModels #Transformers #ProteinImputation #CellTypeAnnotation #BatchCorrection #CellCellCommunication #COVID19 #ComputationalBiology #Bioinformatics
5
1,529

#LabelTransfer #CellTypeAnnotation
popV/popular Vote
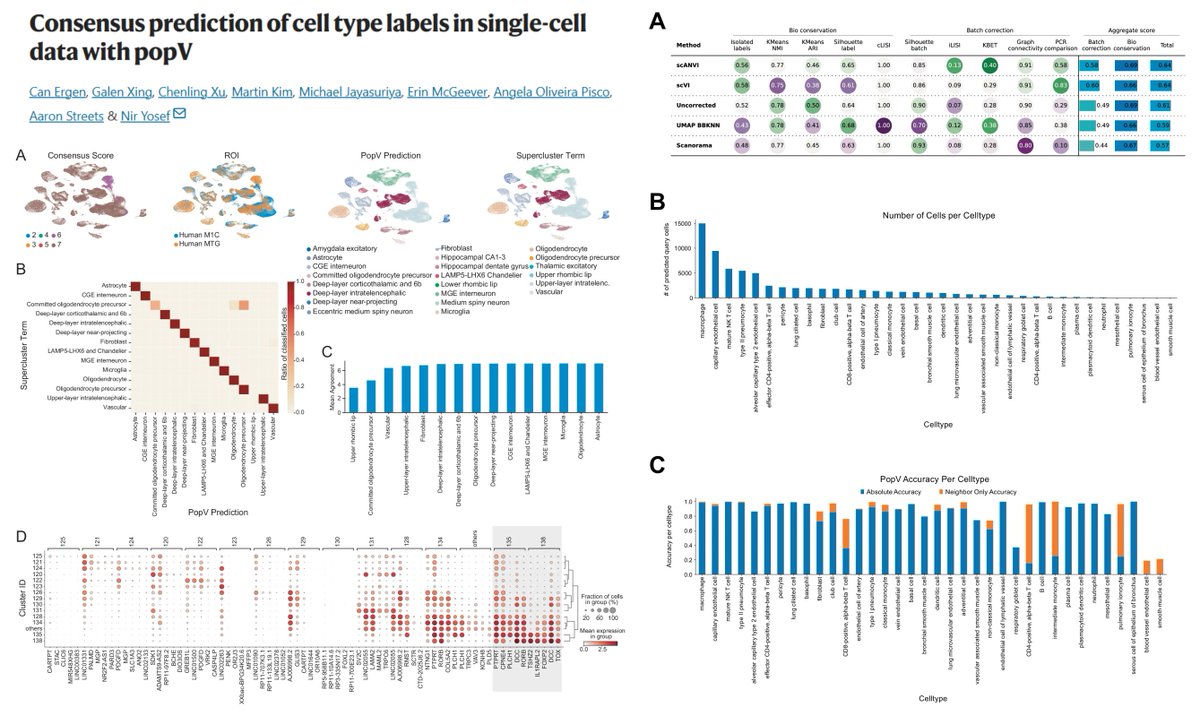
Automated #CellTypeAnnotation Measures of Intrinsic & Extrinsic Uncertainty
Extrinsic: agreement of 8 prediction algorithms
Intrinsic: classifier score of each algorithm
RF SVM Celltypist OnClass KNN Scanorama/BBKNN/scVI/scanVI
Confusion matrices b/w consensus predictions & each method
a Google Colab notebook with Tabula Sapiens 20 organs pre-trained models
"scripts to add custom cell type labels to the Cell Ontology before processing by popV"
Low consensus score/Low accuracy▶️manual annotation, likely needed for most states of #EndothelialCell & #SmoothMuscleCell 🧐
@nir_yosef1 @NatureGenet 2024
nature.com/articles/s41588-0…
14
72
6,769
#LabelTransfer #CellTypeAnnotation
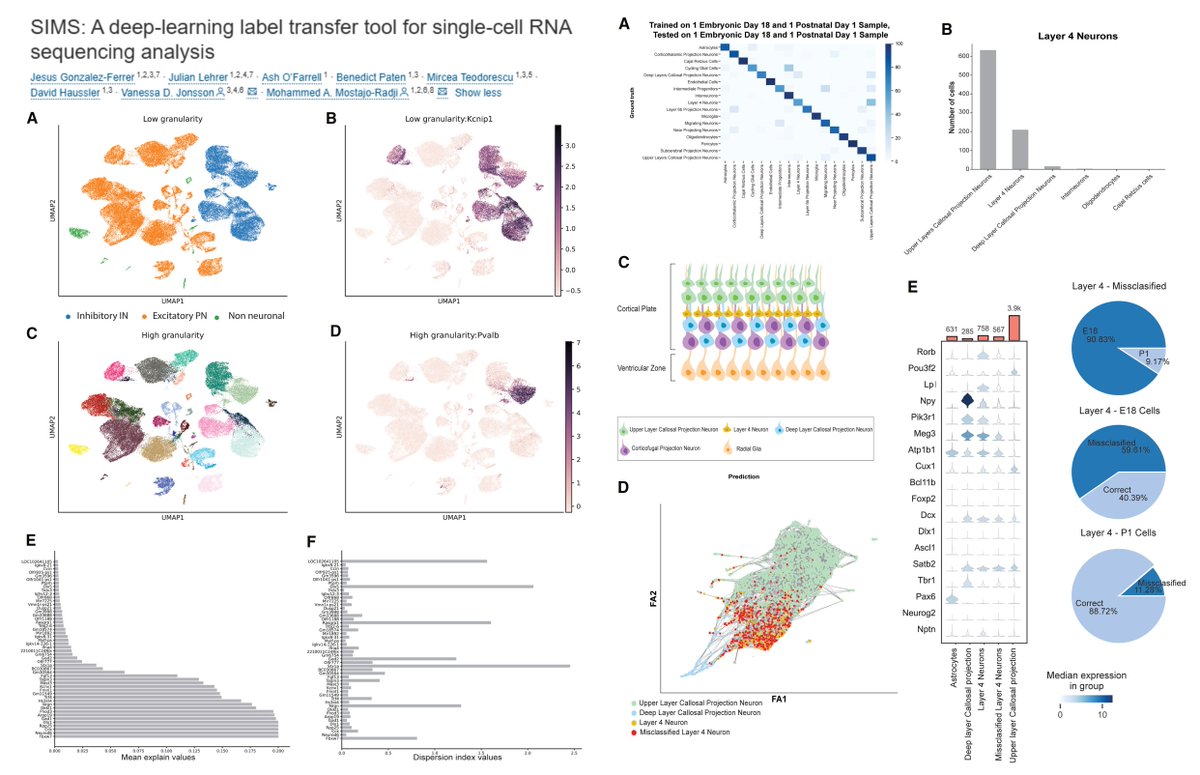
SIMS
Scalable, Interpretable Machine learning for Single cell
github.com/braingeneers/SIMS
TabNet attention-based
Temperature scaling of trained network
Automated gene intersection
vs Seurat scANVI scVI Scnym Scibet SingleR scBERT
"When applied to cortical organoids, SIMS identified previously misannotated cells in existing atlases"
Vanessa Jonsson & Mohammed Mostajo-Radji labs @CellGenomics 2025
cell.com/cell-genomics/fullt…
5
16
1,237

🚀 Excited to share our latest protocol in @NatureProtocols : "scGPT: End-to-end Protocol for Fine-tuned Retinal Cell Type Annotation"!
Single-cell RNA-seq is transforming biology, but annotating large-scale, high-resolution data is challenging. Our team introduces a powerful solution: fine-tuning scGPT, a generative pretrained transformer, specifically optimized for precise cell-type classification in complex datasets.
✨ Key Highlights:
Achieved outstanding performance with 99.5% F1-score on retinal cell annotations.
Comprehensive, easy-to-follow guide covering data preprocessing, fine-tuning, and evaluation.
Streamlined workflow suitable for researchers with minimal programming background.
Includes open-source code, command-line tools, and Jupyter notebooks for quick setup and reproducibility.
🛠️ scGPT significantly simplifies annotation tasks compared to traditional methods (Seurat, scPred, scArches, Geneformer), especially for large datasets and rare cell populations.
💻 Our protocol leverages transformer architectures, harnessing the power of attention mechanisms to capture intricate gene-expression patterns, offering unmatched scalability and precision.
🔗 Check out the full protocol here: doi.org/10.1038/s41596-025-0…
🙏 Huge thanks to our fantastic team: Shanli Ding, Jin Li, Rui Luo, Haotian Cui, Rui Chen, and all collaborators!
#scRNAseq #singlecell #FoundationModels #AI #Transformers #Bioinformatics #CellTypeAnnotation #OpenScience

1
39
206
18,309

1 Jul 2025
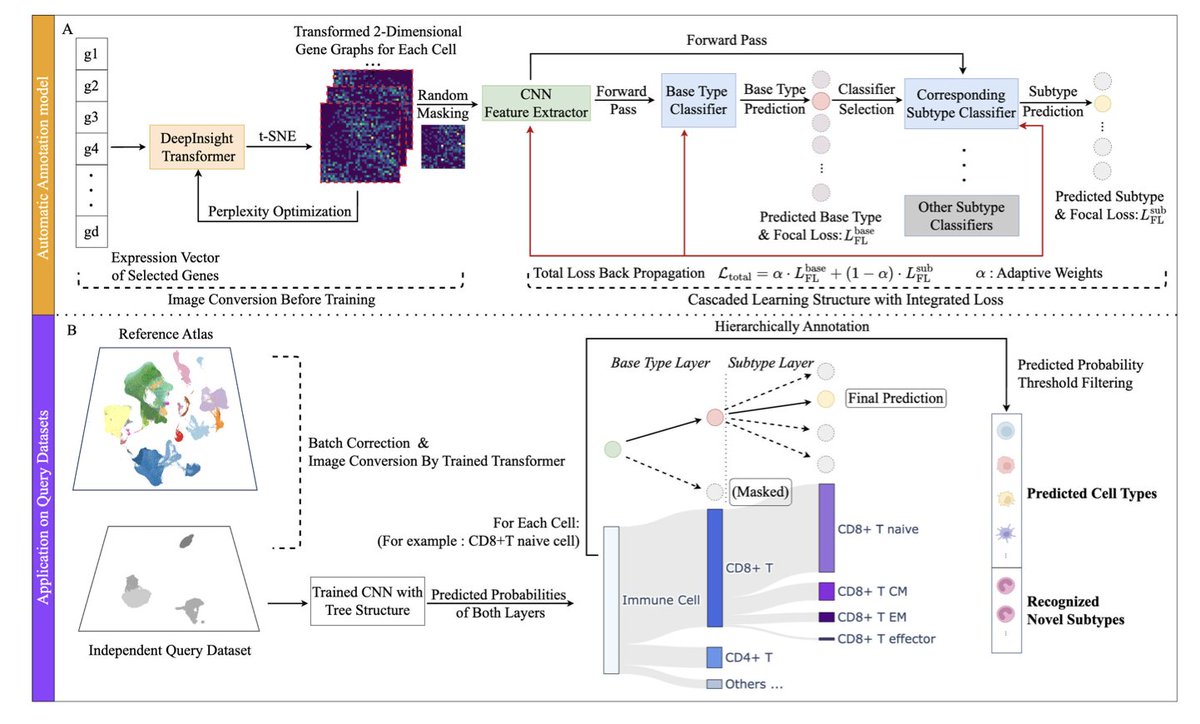
scHDeepInsight: A Hierarchical Deep Learning Framework for Precise Immune Cell Annotation in Single-Cell RNA-seq Data
1.scHDeepInsight introduces a novel hierarchical deep learning framework for immune cell annotation in scRNA-seq, achieving an average accuracy of 93.2% across seven diverse tissue datasets. It significantly outperforms previous state-of-the-art models, with a 5.1% boost in accuracy compared to scDeepInsight.
2.Unlike traditional flat classifiers, scHDeepInsight preserves the biological hierarchy of immune cells. It uses a two-level prediction strategy—first predicting broad immune cell types (base-types), then refining predictions to specific subtypes—reflecting known lineage relationships.
3.The method converts gene expression profiles into structured 2D images using DeepInsight, allowing convolutional neural networks (CNNs) to extract both global and fine-grained transcriptomic patterns. This spatial representation helps capture complex gene-gene relationships.
4.A key innovation is the Adaptive Hierarchical Focal Loss (AHFL), which dynamically balances the classification loss at the base-type and subtype levels, adapting training focus based on task difficulty and addressing class imbalance in rare subtypes.
5.scHDeepInsight incorporates STACAS for batch effect correction and applies random masking during training to improve robustness against missing gene features, enabling cross-dataset generalization.
6.The framework includes SHAP-based interpretability, quantifying the contribution of each gene to classification decisions at both base and subtype levels. This reveals both canonical markers (like CD8A for CD8 T cells) and subtle subtype-specific signatures (e.g., IGHA1 for IgA plasma cells).
7.It effectively identifies rare and novel immune cell populations. In glioblastoma data, the model detected glioma-associated immune cells by recognizing high base-type confidence but low subtype confidence—suggesting novel states outside the training reference.
8.Comprehensive benchmarking shows that scHDeepInsight consistently outperforms methods like SingleR, Azimuth, CellTypist, GPTCellType, and Garnett across accuracy, precision, F1-score, and AUPRC—even for challenging closely related immune subtypes.
9.In specific case studies (e.g., Pranzatelli labial gland dataset), scHDeepInsight distinguished IgA , IgG , and IgM plasma cell subtypes that were grouped together by other models, highlighting its resolution and biological fidelity.
10.The reference atlas used to train the model includes over 460,000 immune cells from 10 public scRNA-seq datasets, spanning 15 major immune lineages and 50 subtypes. This comprehensive dataset provides a robust foundation for hierarchical learning.
11.Future directions for scHDeepInsight include extending the hierarchy to non-immune cells, incorporating multi-omics (e.g., CITE-seq, spatial transcriptomics), and applying self-supervised or transfer learning to improve adaptability to new datasets or species.
💻Code: github.com/shangruJia/scHDee…
📜Paper: doi.org/10.1101/2025.06.23.6…
#scRNAseq #DeepLearning #CellTypeAnnotation #Immunology #SingleCell #CNN #Bioinformatics
3
831
1 Jul 2025
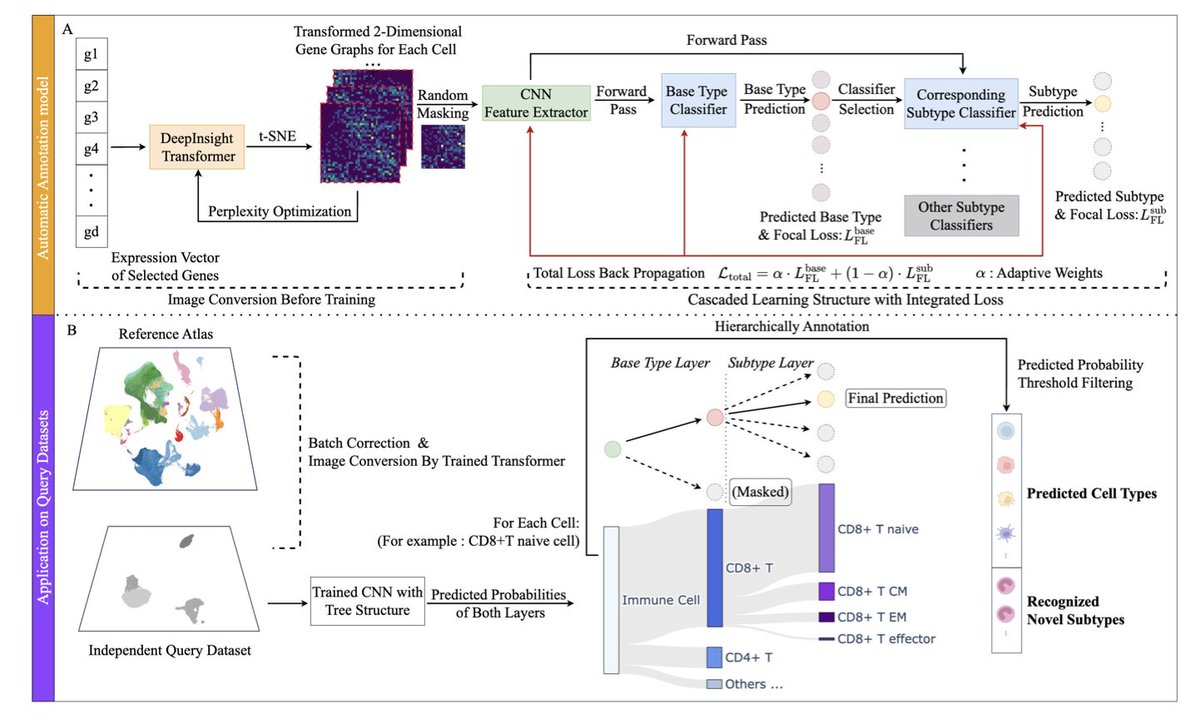
scHDeepInsight: A Hierarchical Deep Learning Framework for Precise Immune Cell Annotation in Single-Cell RNA-seq Data
1.scHDeepInsight introduces a novel hierarchical deep learning framework for immune cell annotation in scRNA-seq, achieving an average accuracy of 93.2% across seven diverse tissue datasets. It significantly outperforms previous state-of-the-art models, with a 5.1% boost in accuracy compared to scDeepInsight.
2.Unlike traditional flat classifiers, scHDeepInsight preserves the biological hierarchy of immune cells. It uses a two-level prediction strategy—first predicting broad immune cell types (base-types), then refining predictions to specific subtypes—reflecting known lineage relationships.
3.The method converts gene expression profiles into structured 2D images using DeepInsight, allowing convolutional neural networks (CNNs) to extract both global and fine-grained transcriptomic patterns. This spatial representation helps capture complex gene-gene relationships.
4.A key innovation is the Adaptive Hierarchical Focal Loss (AHFL), which dynamically balances the classification loss at the base-type and subtype levels, adapting training focus based on task difficulty and addressing class imbalance in rare subtypes.
5.scHDeepInsight incorporates STACAS for batch effect correction and applies random masking during training to improve robustness against missing gene features, enabling cross-dataset generalization.
6.The framework includes SHAP-based interpretability, quantifying the contribution of each gene to classification decisions at both base and subtype levels. This reveals both canonical markers (like CD8A for CD8 T cells) and subtle subtype-specific signatures (e.g., IGHA1 for IgA plasma cells).
7.It effectively identifies rare and novel immune cell populations. In glioblastoma data, the model detected glioma-associated immune cells by recognizing high base-type confidence but low subtype confidence—suggesting novel states outside the training reference.
8.Comprehensive benchmarking shows that scHDeepInsight consistently outperforms methods like SingleR, Azimuth, CellTypist, GPTCellType, and Garnett across accuracy, precision, F1-score, and AUPRC—even for challenging closely related immune subtypes.
9.In specific case studies (e.g., Pranzatelli labial gland dataset), scHDeepInsight distinguished IgA , IgG , and IgM plasma cell subtypes that were grouped together by other models, highlighting its resolution and biological fidelity.
10.The reference atlas used to train the model includes over 460,000 immune cells from 10 public scRNA-seq datasets, spanning 15 major immune lineages and 50 subtypes. This comprehensive dataset provides a robust foundation for hierarchical learning.
11.Future directions for scHDeepInsight include extending the hierarchy to non-immune cells, incorporating multi-omics (e.g., CITE-seq, spatial transcriptomics), and applying self-supervised or transfer learning to improve adaptability to new datasets or species.
💻Code: github.com/shangruJia/scHDee…
📜Paper: biorxiv.org/content/10.1101/…
#scRNAseq #DeepLearning #CellTypeAnnotation #Immunology #SingleCell #CNN #Bioinformatics
2
544
6 Apr 2025
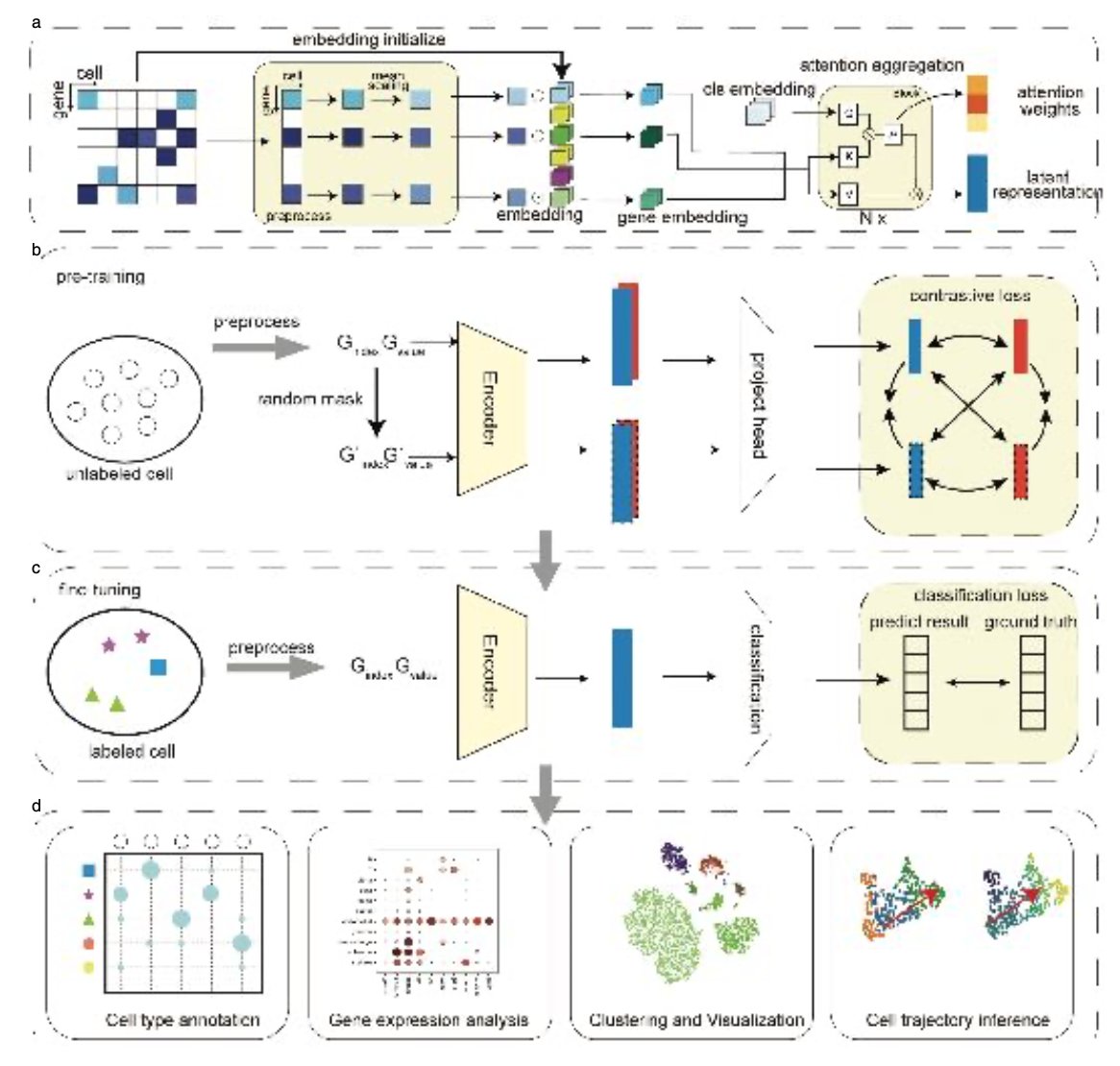
scTrans: Sparse attention powers fast and accurate cell type annotation in single-cell RNA-seq data
1. scTrans introduces a sparse-attention Transformer framework that enables fast, accurate, and interpretable cell type annotation in single-cell RNA-seq datasets, even at million-cell scale and under limited hardware resources.
2. Unlike traditional methods relying on highly variable genes (HVGs), scTrans utilizes all non-zero gene expressions via sparse attention, reducing dimensionality while minimizing information loss.
3. scTrans significantly outperforms existing methods across 31 mouse tissues in the Mouse Cell Atlas, showing up to 6% higher accuracy and 16% higher F1-macro than the next-best method with only 10% labeled data.
4. On ultra-large datasets like PBMC160k (161k cells) and scBloodNL (928k cells), scTrans delivers top-tier accuracy while consuming the least runtime and memory—running efficiently within 40GB RAM.
5. A lightweight variant, scTrans-short, retains competitive performance while further reducing runtime, demonstrating the model’s scalability and practicality for large-scale projects.
6. The model exhibits robust generalization to novel datasets and is resilient to batch effects, enabling accurate cross-batch annotations without retraining or requiring batch information.
7. Attention weights in scTrans are biologically informative, helping identify critical or marker genes and uncover functionally relevant pathways via enrichment analyses.
8. scTrans achieves high-quality latent representations ideal for downstream tasks like clustering and trajectory inference, outperforming both supervised and unsupervised alternatives across ARI, NMI, and ASW metrics.
9. In trajectory analyses, scTrans accurately reconstructs developmental paths for human T cells and mouse dendritic cells, aligning inferred pseudotime with known gene expression dynamics.
10. The model successfully distinguishes subtle subtypes within cell populations, such as naive vs memory B cells and dendritic cell subtypes, validated by expression of canonical markers like IGHD, TCL1A, and CLEC10A.
11. Compared with large foundation models like scGPT and CellPLM, scTrans achieves better performance in detailed annotation tasks and retains superior sensitivity to fine cell-type distinctions.
12. The codebase is efficient, modular, and interpretable, enabling both expert and non-expert users to deploy scTrans on diverse datasets without GPU-heavy environments.
💻Code: github.com/zou-zhiyi/scTrans
📜Paper: journals.plos.org/ploscompbi…
#SingleCell #scRNAseq #DeepLearning #Transformer #Bioinformatics #CellTypeAnnotation #SparseAttention #ComputationalBiology #TrajectoryInference #GeneExpression
1
6
1,183
14 Feb 2025
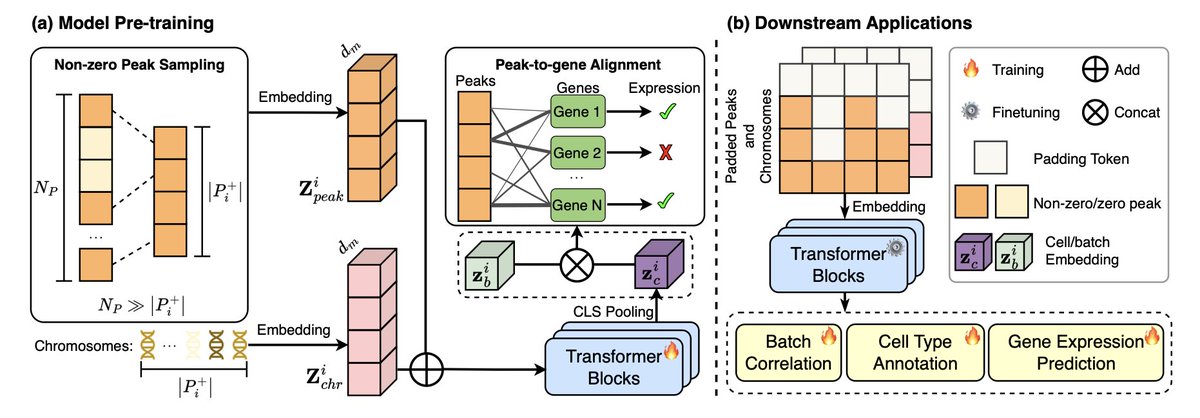
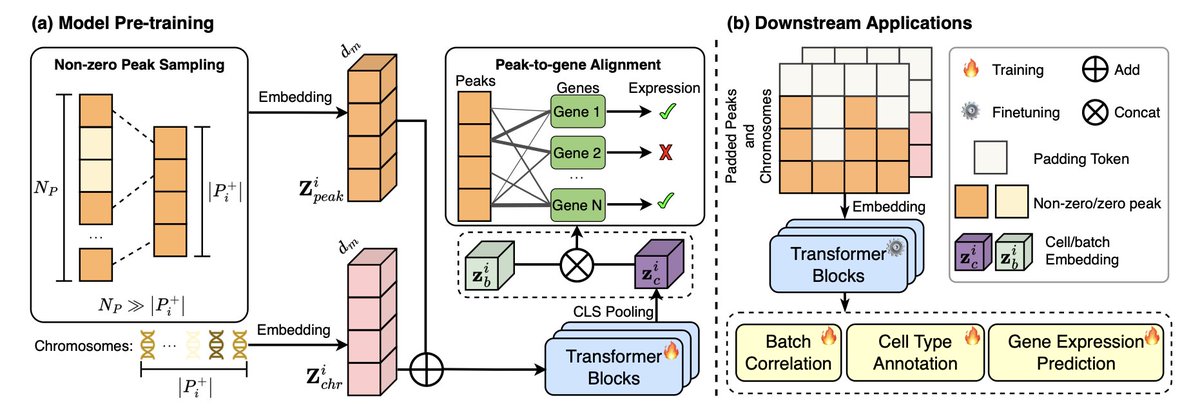
EpiFoundation: A Foundation Model for Single-Cell ATAC-seq via Peak-to-Gene Alignment
1/ EpiFoundation is a novel foundation model designed for single-cell ATAC-seq data, addressing the challenges of high-dimensional, sparse peak data. It uses innovative peak-to-gene alignment to enhance model accuracy in cell representation.
2/ The model processes only non-zero peaks in single cells, significantly improving computational efficiency and cell-specific representation. This approach contrasts with previous methods that often include all peaks, regardless of their expression status.
3/ EpiFoundation employs a cross-modality pre-training strategy, aligning ATAC-seq peaks with gene expression data, to better correlate chromatin accessibility with gene activity, ensuring robust and meaningful cell representation.
4/ The model is trained using the MiniAtlas dataset, which includes over 100,000 cells across 19 tissues and 56 cell types, enabling EpiFoundation to handle diverse tissue types and cell states for multiple downstream applications like cell-type annotation and gene expression prediction.
5/ EpiFoundation outperforms existing methods in several tasks, including cell type annotation, batch effect correction, and gene expression prediction. It achieves superior performance metrics, such as higher accuracy and improved biological conservation during batch correction.
6/ For gene expression prediction, EpiFoundation significantly surpasses other methods like Gene Activity, demonstrating its power in accurately linking peak expression data to gene activity and improving prediction precision.
7/ The model’s robustness across diverse biological datasets makes it a versatile tool for single-cell epigenomic research, offering a new way to integrate ATAC-seq and RNA-seq data for better understanding gene regulation in health and disease.
💻Code: github.com/UCSC-VLAA/EpiFoun…
📜Paper: biorxiv.org/content/10.1101/…
#SingleCell #ATACseq #GeneExpression #Bioinformatics #MachineLearning #Epigenomics #BatchCorrection #CellTypeAnnotation #AIinBiology #ComputationalBiology
3
11
1,373
14 Feb 2025
EpiFoundation: A Foundation Model for Single-Cell ATAC-seq via Peak-to-Gene Alignment
1/ EpiFoundation is a novel foundation model designed for single-cell ATAC-seq data, addressing the challenges of high-dimensional, sparse peak data. It uses innovative peak-to-gene alignment to enhance model accuracy in cell representation.
2/ The model processes only non-zero peaks in single cells, significantly improving computational efficiency and cell-specific representation. This approach contrasts with previous methods that often include all peaks, regardless of their expression status.
3/ EpiFoundation employs a cross-modality pre-training strategy, aligning ATAC-seq peaks with gene expression data, to better correlate chromatin accessibility with gene activity, ensuring robust and meaningful cell representation.
4/ The model is trained using the MiniAtlas dataset, which includes over 100,000 cells across 19 tissues and 56 cell types, enabling EpiFoundation to handle diverse tissue types and cell states for multiple downstream applications like cell-type annotation and gene expression prediction.
5/ EpiFoundation outperforms existing methods in several tasks, including cell type annotation, batch effect correction, and gene expression prediction. It achieves superior performance metrics, such as higher accuracy and improved biological conservation during batch correction.
6/ For gene expression prediction, EpiFoundation significantly surpasses other methods like Gene Activity, demonstrating its power in accurately linking peak expression data to gene activity and improving prediction precision.
7/ The model’s robustness across diverse biological datasets makes it a versatile tool for single-cell epigenomic research, offering a new way to integrate ATAC-seq and RNA-seq data for better understanding gene regulation in health and disease.
@HWenpin @yuyinzhou_cs @ZhichengJi @Changson_Wan
💻Code: github.com/UCSC-VLAA/EpiFoun…
📜Paper: biorxiv.org/content/10.1101/…
#SingleCell #ATACseq #GeneExpression #Bioinformatics #MachineLearning #Epigenomics #BatchCorrection #CellTypeAnnotation #AIinBiology #ComputationalBiology
4
6
998
14 Feb 2025
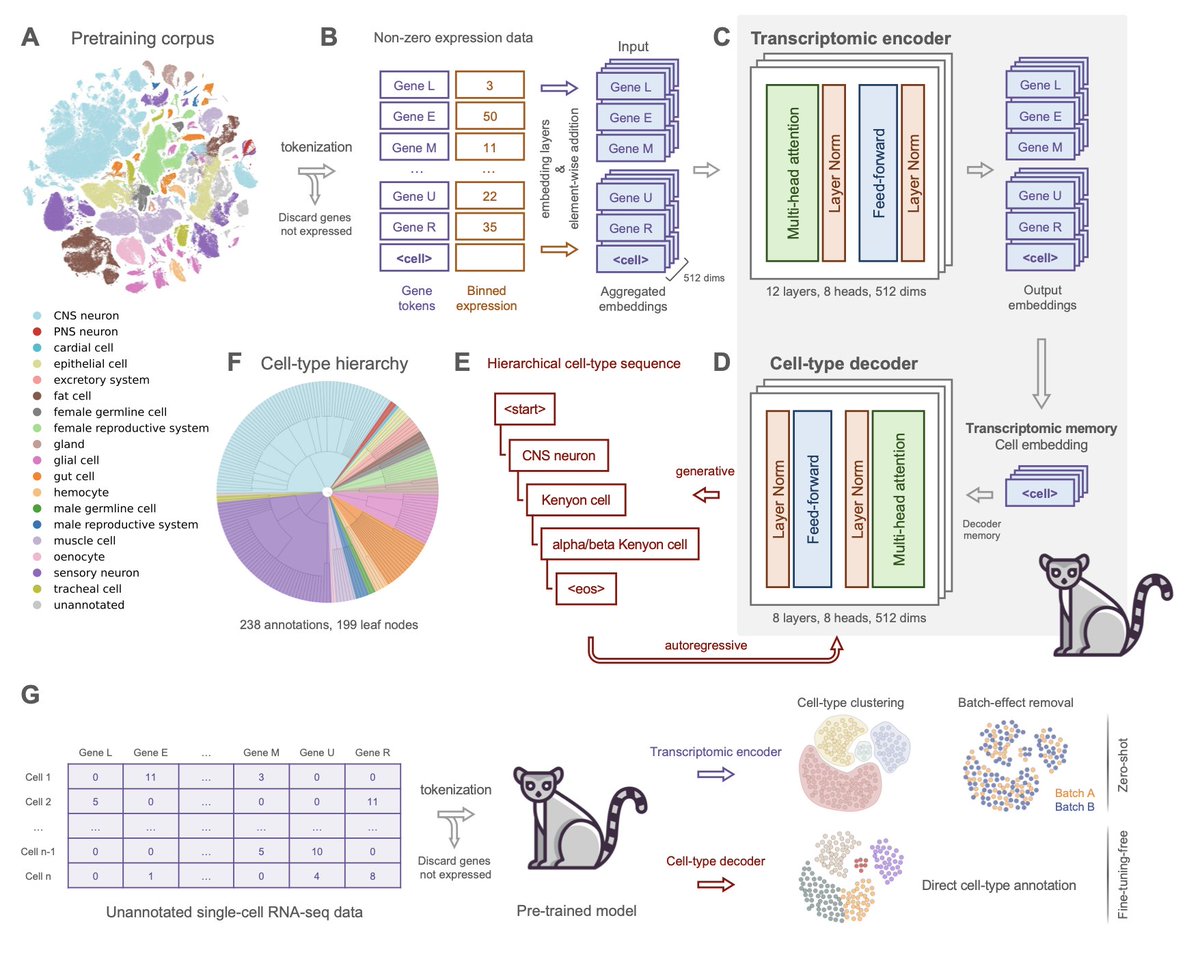
Lemur: A Single-Cell Foundation Model with Fine-Tuning-Free Hierarchical Cell-Type Generation for Drosophila melanogaster
1/ Lemur is a novel single-cell foundation model designed for Drosophila melanogaster, offering a fine-tuning-free approach for accurate cell-type annotation, using a hierarchical cell-type decoder to achieve precise labeling across various tissues and experimental conditions.
2/ The model was pre-trained on a comprehensive Drosophila single-nucleus RNA-seq dataset, integrating multiple atlases to create a unified cell-type annotation schema, which enables Lemur to classify cells without requiring additional training on new datasets.
3/ Lemur’s hierarchical decoding mechanism allows it to predict cell-type labels across various levels of granularity, from broad categories like CNS neurons to specific subtypes, ensuring biological consistency even in complex tissue types where fine-grained annotations are challenging.
4/ The model’s fine-tuning-free nature is a significant advantage, making it ready for use across diverse datasets without needing further model adaptation, which streamlines workflows in single-cell genomic analysis.
5/ Lemur demonstrates strong batch-effect correction performance without explicit training for the task, providing a zero-shot solution that enhances data integration while preserving biological signals, outperforming specialized batch correction methods like scANVI.
6/ Evaluations across wild-type and disease models, including Alzheimer's and Parkinson’s, show that Lemur maintains robust performance with high precision and comprehensive cell-type coverage, demonstrating its potential for broad applications in developmental biology and neurodegenerative disease research.
7/ This efficient, scalable tool has the potential to accelerate discoveries in cellular heterogeneity, aging, and disease, particularly by enabling rapid iteration between computational predictions and experimental validation in the Drosophila system.
💻Code: huggingface.co/jorgebotas/le…
📜Paper: biorxiv.org/content/10.1101/…
#SingleCell #Bioinformatics #MachineLearning #AIinBiology #Drosophila #CellTypeAnnotation #ComputationalBiology #BatchEffectCorrection #NeurodegenerativeDiseases #SingleCellGenomics
1
8
1,057
14 Feb 2025
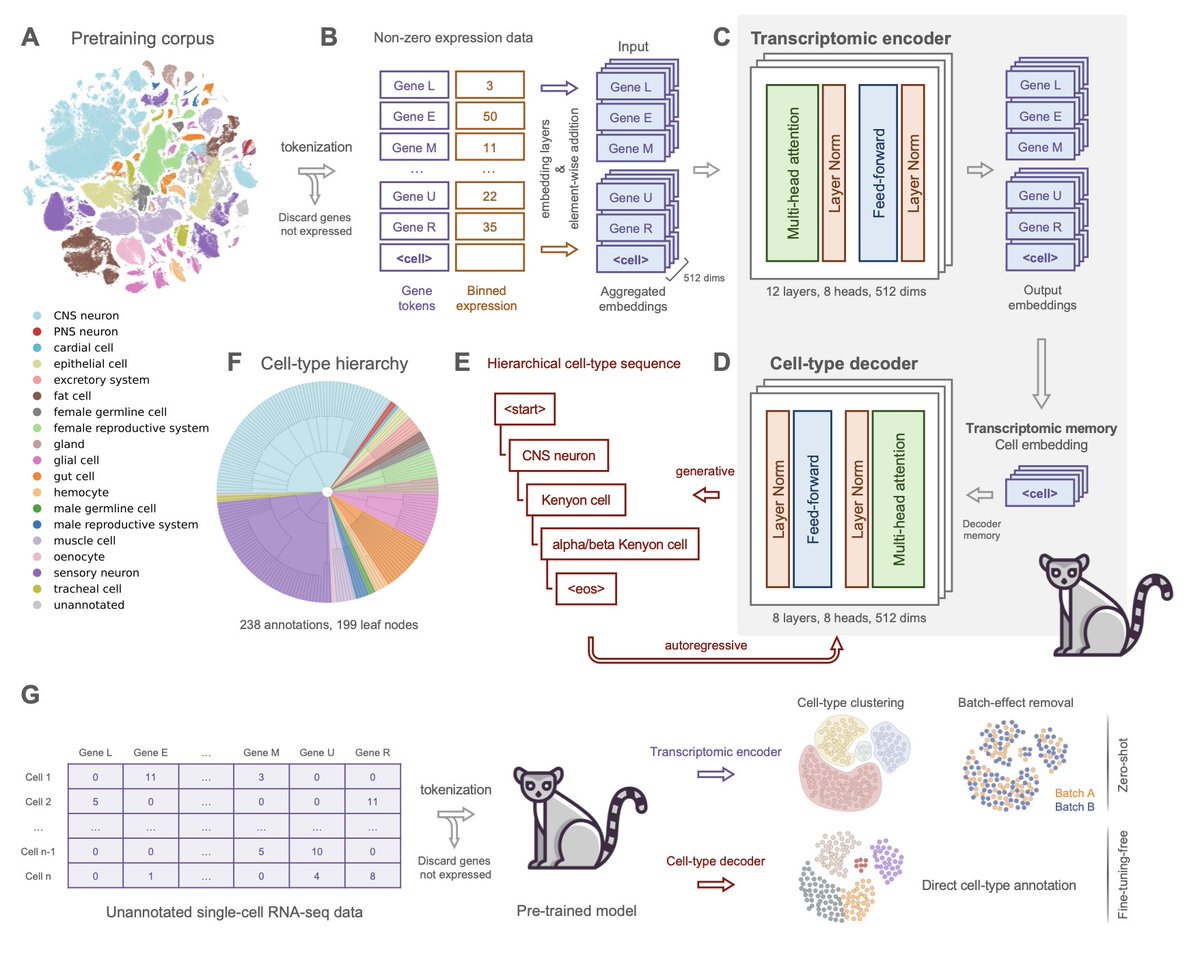
Lemur: A Single-Cell Foundation Model with Fine-Tuning-Free Hierarchical Cell-Type Generation for Drosophila melanogaster
1/ Lemur is a novel single-cell foundation model designed for Drosophila melanogaster, offering a fine-tuning-free approach for accurate cell-type annotation, using a hierarchical cell-type decoder to achieve precise labeling across various tissues and experimental conditions.
2/ The model was pre-trained on a comprehensive Drosophila single-nucleus RNA-seq dataset, integrating multiple atlases to create a unified cell-type annotation schema, which enables Lemur to classify cells without requiring additional training on new datasets.
3/ Lemur’s hierarchical decoding mechanism allows it to predict cell-type labels across various levels of granularity, from broad categories like CNS neurons to specific subtypes, ensuring biological consistency even in complex tissue types where fine-grained annotations are challenging.
4/ The model’s fine-tuning-free nature is a significant advantage, making it ready for use across diverse datasets without needing further model adaptation, which streamlines workflows in single-cell genomic analysis.
5/ Lemur demonstrates strong batch-effect correction performance without explicit training for the task, providing a zero-shot solution that enhances data integration while preserving biological signals, outperforming specialized batch correction methods like scANVI.
6/ Evaluations across wild-type and disease models, including Alzheimer's and Parkinson’s, show that Lemur maintains robust performance with high precision and comprehensive cell-type coverage, demonstrating its potential for broad applications in developmental biology and neurodegenerative disease research.
7/ This efficient, scalable tool has the potential to accelerate discoveries in cellular heterogeneity, aging, and disease, particularly by enabling rapid iteration between computational predictions and experimental validation in the Drosophila system.
@ZhandongLiu @ZhaozhuoX
💻Code: huggingface.co/jorgebotas/le…
📜Paper: biorxiv.org/content/10.1101/…
#SingleCell #Bioinformatics #MachineLearning #AIinBiology #Drosophila #CellTypeAnnotation #ComputationalBiology #BatchEffectCorrection #NeurodegenerativeDiseases #SingleCellGenomics
4
7
1,433

10 Nov 2024
scATAcat: cell-type annotation for scATAC-seq data. #scATACseq #CellTypeAnnotation #NARgenomicsAndBioinformatics
academic.oup.com/nargab/arti…
1
3
470
14 Mar 2023
scAnnotate: an automated cell type annotation tool for single-cell RNA-sequencing data. #scRNAseq #CellTypeAnnotation @BioinfoAdv academic.oup.com/bioinformat…
2
13
851

8 Sep 2021
Single-Cell Transcriptome Profiling Simulation Reveals the Impact of Sequencing Parameters and Algorithms on Clustering. View Full-Text: mdpi.com/2075-1729/11/7/716
@MissionBio
#bioinformatics; #clustering; #celltypeannotation
3
3
25 May 2021
CDSeqR: fast complete deconvolution for gene expression data from bulk tissues. #RNAseq #GeneExpression #CDSeq #Rpackage #CellTypeAnnotation #Bioinformatics
bmcbioinformatics.biomedcent… @BMCBioinfo
1
1