Fast sampling of protein conformational dynamics @ScienceAdvances
1. Sauer et al. show that the key collective variables (CVs) needed to drive enhanced sampling of protein conformational transitions are encoded in anharmonic low-frequency vibrations, and these CVs can be extracted from short unbiased MD without any prior knowledge of the transition.
2. Core idea: use FRESEAN (frequency-selective anharmonic mode analysis) at (near) zero frequency to isolate collective motions with minimal restoring forces—i.e., “paths of least resistance” for conformational change—avoiding the limitations of harmonic/quasiharmonic normal modes in the low-frequency, diffusive regime.
3. Practical pipeline: run 20 ns unbiased all-atom MD, align trajectories, coarse-grain to a 2-bead-per-residue representation (1 for Gly), compute velocity time-correlation matrices, Fourier transform to frequency domain, then take eigenvectors at zero frequency. Modes 1–6 correspond to translation/rotation and are discarded; modes 7 capture internal anharmonic low-frequency vibrations.
4. Reproducibility is a central result: across 5 independent 20 ns replicas per protein, the low-frequency modes (especially the 2D subspace spanned by modes 7–8) are consistently recovered, unlike PCA/quasiharmonic modes whose replica-to-replica agreement remains poor even with much longer trajectories.
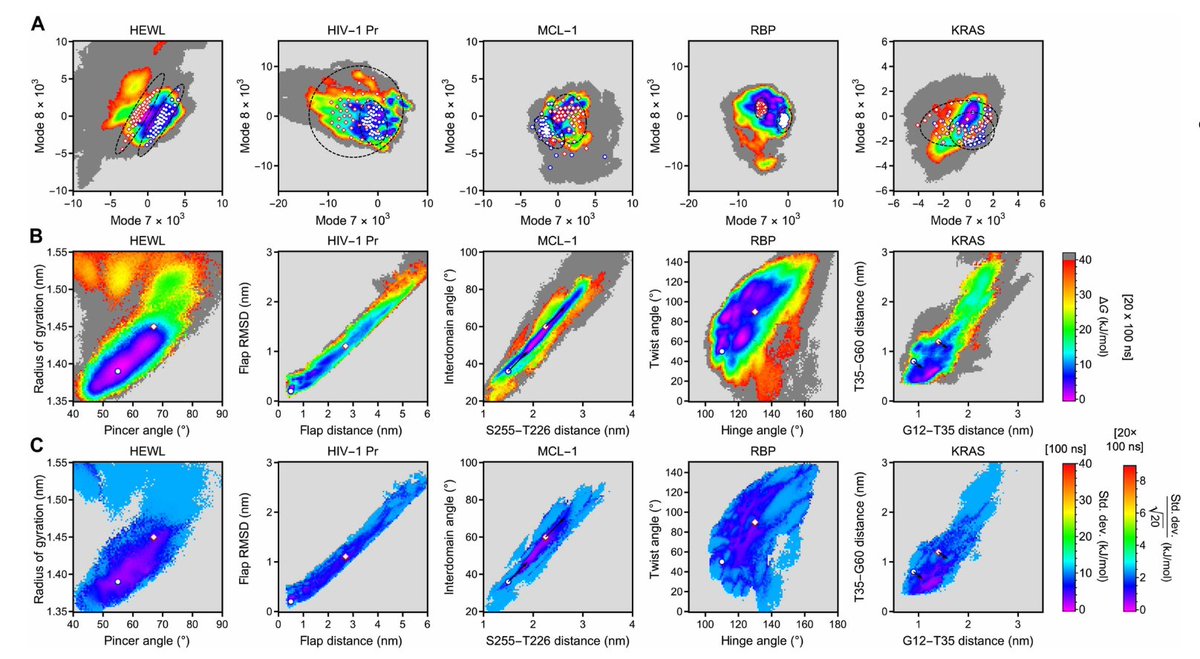
5. Enhanced sampling step: use modes 7 and 8 as CVs in well-tempered metadynamics (100 ns per run; reported as <24 hours on a single GPU). Across 5 proteins × 5 replicas, 22/25 runs (88%) sample known “closed↔open” transitions within 100 ns; extending to 160 ns yields full sampling for all replicas.
6. Benchmark set spans diverse challenges: HEWL (disulfide-stabilized), HIV-1 protease (homodimer), MCL-1 (allosteric/druggable dynamics), ribose-binding protein (multi-domain hinge motion), and GDP-bound KRAS (switch-region dynamics). The same FRESEAN-to-metadynamics protocol is applied across all systems.
7. Free-energy landscapes (FES) become both fast and statistically controlled by running 20 parallel metadynamics replicas (20 × 100 ns) using the same FRESEAN CVs: single-run uncertainties are typically < ±10 kJ/mol, and averaging reduces standard error to < ±3 kJ/mol, enabling reproducible thermodynamic ensembles rather than just qualitative transitions.
8. Comparison to “hand-crafted” geometric CVs from prior literature is informative: biasing along FRESEAN modes often follows lower-free-energy transition routes and tends to keep sampling within the native folded ensemble, whereas geometric CVs can push systems into partially unfolded high-entropy states (most notably KRAS when biased by residue–residue distances).
9. The authors quantify cross-CV reweighting fidelity using Shannon entropy and Bhattacharyya coefficients: on average, ensembles generated by biasing along low-frequency vibrational CVs preserve at least as much (often more) information when reweighted into geometric-variable space than the reverse, supporting the claim that these vibrations are broadly suitable, system-agnostic CVs.
10. Implication for computational biology/ML: the method enables high-throughput generation of conformational ensembles and FESs (including mutants/conditions), helping address the dataset bottleneck for next-generation sequence→structure→dynamics models beyond single static folds or single thermodynamic states.
💻Code: github.com/HeydenLabASU-coll…
📜Paper: doi.org/10.1126/sciadv.aea46…
#MolecularDynamics #EnhancedSampling #Metadynamics #ProteinDynamics #FreeEnergy #ComputationalBiophysics #CollectiveVariables #FRESEAN #GROMACS #PLUMED
16
70
4,180

16 Oct 2025
Enhancing Diffusion-Based Sampling with Molecular Collective Variables
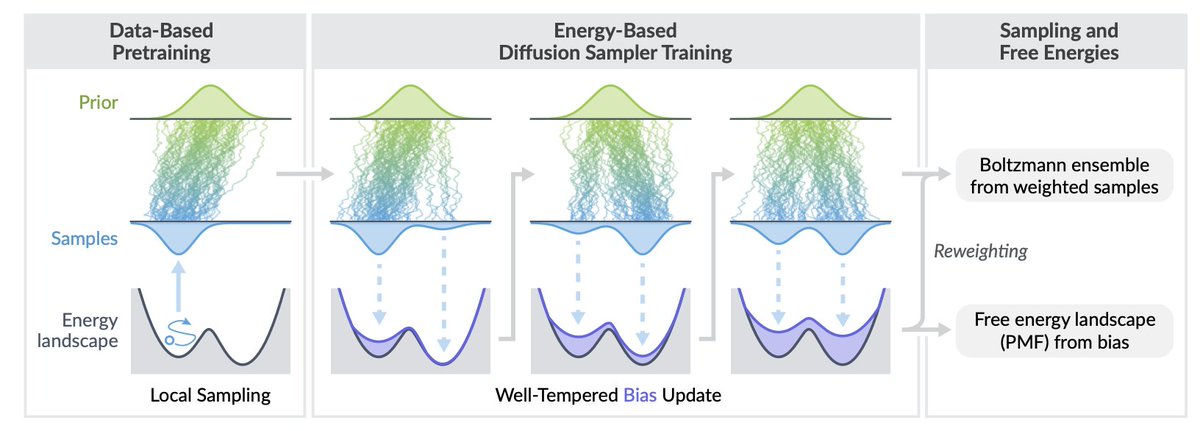
1. A new method called Well-Tempered Adjoint Schrödinger Bridge Sampler (WT-ASBS) is introduced to improve molecular sampling efficiency. This method combines diffusion-based sampling with a well-tempered bias in collective variable (CV) space, enabling more efficient exploration of molecular configurations and accurate estimation of free energy differences.
2. The WT-ASBS method addresses the limitations of traditional diffusion-based samplers, which often suffer from slow sampling speeds and mode collapse. By introducing a repulsive potential centered on CVs from recent samples, it pushes future samples towards novel CV regions, effectively increasing the temperature in the projected space and enhancing exploration.
3. The method is demonstrated on peptide conformational sampling benchmarks, where it recovers diverse conformational states and accurate free energy profiles. It is also the first to demonstrate reactive sampling using a diffusion-based sampler, capturing bond breaking and formation with universal interatomic potentials at near-first-principles accuracy.
4. WT-ASBS achieves significant improvements in sampling efficiency compared to traditional methods like well-tempered metadynamics (WTMetaD). For example, in sampling reactive landscapes involving bond formation, WT-ASBS required significantly fewer energy evaluations and shorter wall-clock times than WTMetaD.
5. The approach resolves reactive energy landscapes at a fraction of the wall-clock time of standard sampling methods, advancing diffusion-based sampling towards practical use in molecular sciences. This opens up new possibilities for studying complex molecular systems and reactions with high accuracy and efficiency.
📜Paper: arxiv.org/abs/2510.11923
#MolecularSampling #DiffusionBasedSampling #CollectiveVariables #FreeEnergy #ReactiveLandscapes
7
52
2,736
23 May 2025
NeuralTSNE: A Python Package for the Dimensionality Reduction of Molecular Dynamics Data Using Neural Networks
1.NeuralTSNE is a new Python package implementing parametric t-SNE using neural networks for dimensionality reduction of molecular dynamics (MD) data. It enables high-fidelity mapping of complex molecular trajectories into low-dimensional representations ideal for analysis and visualization.
2.Unlike standard t-SNE, NeuralTSNE uses a neural network to learn a continuous mapping from high-dimensional feature space to low-dimensional latent space. This allows reusability of the trained model on new data—a key advantage for MD workflows.
3.The algorithm minimizes the Kullback-Leibler divergence between high-dimensional and reduced-space distance distributions using a feedforward neural network trained via PyTorch and PyTorch Lightning. It supports GPU acceleration and is easy to integrate into modern ML pipelines.
4.NeuralTSNE is designed for usability: it can be run as a command-line tool or imported as a Python module. Its API is consistent with scikit-learn’s interface, providing familiar fit and predict functions.
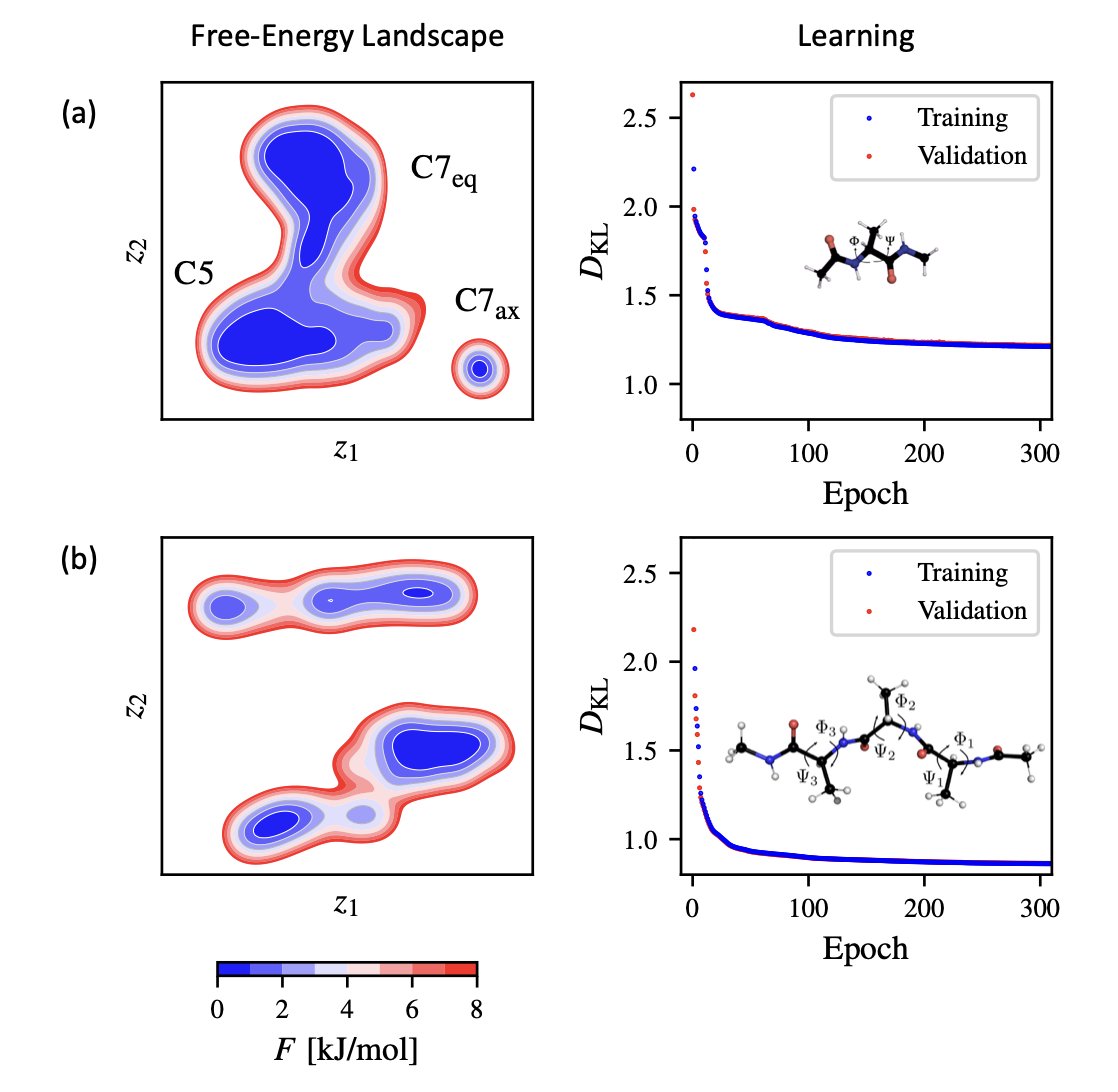
5.The package is demonstrated on MD datasets from alanine dipeptide and tetrapeptide simulations. It successfully reconstructs known free-energy landscapes, recovering metastable states and transition barriers with remarkable precision.
6.In the alanine dipeptide case, NeuralTSNE identifies three key states—C7eq, C5, and C7ax—matching established dihedral-based mappings. For the tetrapeptide, the model identifies complex multi-state landscapes, highlighting its capacity to handle high-dimensional, rugged data.
7.Unlike static t-SNE implementations, NeuralTSNE generalizes well to out-of-sample data, making it suitable for online analysis and enhanced sampling simulations. The authors plan to integrate it with PLUMED to guide MD trajectories using learned coordinates.
8.The package provides full reproducibility: examples, testing frameworks, CI/CD pipelines, and documentation are available on GitHub. Tutorials cover MD data and general datasets like MNIST, making it accessible for users across domains.
9.NeuralTSNE opens the door to future extensions such as reweighted embeddings, spectral maps, and direct use of the learned coordinates in biasing simulations. It fills a key gap in ML tools for physics-informed MD analysis.
10.This implementation democratizes access to parametric t-SNE for chemists and physicists, offering a practical and extensible tool for understanding molecular behavior beyond intuition-driven reaction coordinates.
💻Code: github.com/NeuralTSNE/Neural… 📜Paper: arxiv.org/abs/2505.16476
#MolecularDynamics #DimensionalityReduction #tSNE #NeuralNetworks #CollectiveVariables #UnsupervisedLearning #ComputationalChemistry #NeuralTSNE #EnhancedSampling
3
31
1,849
23 May 2025
NeuralTSNE: A Python Package for the Dimensionality Reduction of Molecular Dynamics Data Using Neural Networks
1.NeuralTSNE is a new Python package implementing parametric t-SNE using neural networks for dimensionality reduction of molecular dynamics (MD) data. It enables high-fidelity mapping of complex molecular trajectories into low-dimensional representations ideal for analysis and visualization.
2.Unlike standard t-SNE, NeuralTSNE uses a neural network to learn a continuous mapping from high-dimensional feature space to low-dimensional latent space. This allows reusability of the trained model on new data—a key advantage for MD workflows.
3.The algorithm minimizes the Kullback-Leibler divergence between high-dimensional and reduced-space distance distributions using a feedforward neural network trained via PyTorch and PyTorch Lightning. It supports GPU acceleration and is easy to integrate into modern ML pipelines.
4.NeuralTSNE is designed for usability: it can be run as a command-line tool or imported as a Python module. Its API is consistent with scikit-learn’s interface, providing familiar fit and predict functions.
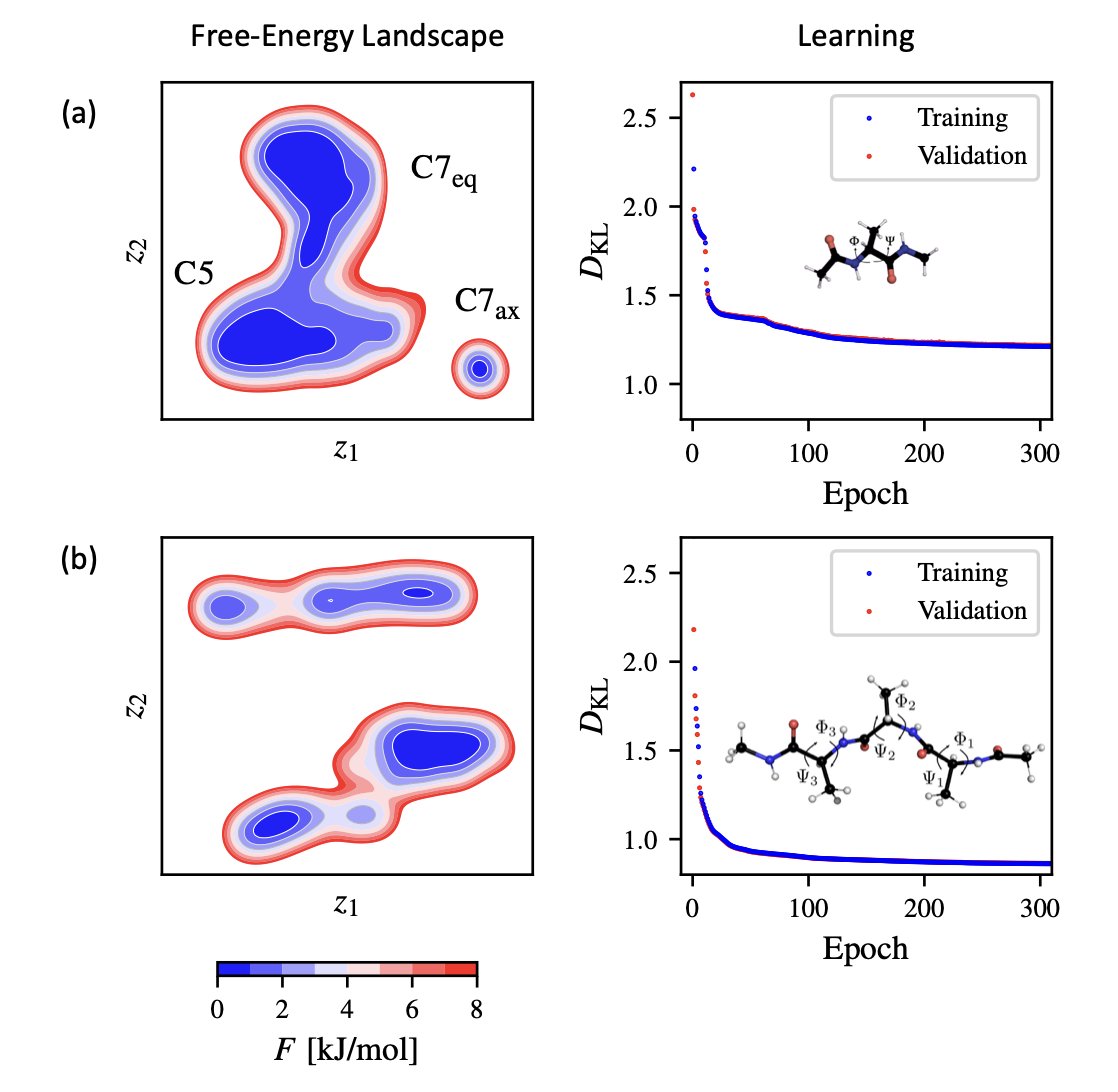
5.The package is demonstrated on MD datasets from alanine dipeptide and tetrapeptide simulations. It successfully reconstructs known free-energy landscapes, recovering metastable states and transition barriers with remarkable precision.
6.In the alanine dipeptide case, NeuralTSNE identifies three key states—C7eq, C5, and C7ax—matching established dihedral-based mappings. For the tetrapeptide, the model identifies complex multi-state landscapes, highlighting its capacity to handle high-dimensional, rugged data.
7.Unlike static t-SNE implementations, NeuralTSNE generalizes well to out-of-sample data, making it suitable for online analysis and enhanced sampling simulations. The authors plan to integrate it with PLUMED to guide MD trajectories using learned coordinates.
8.The package provides full reproducibility: examples, testing frameworks, CI/CD pipelines, and documentation are available on GitHub. Tutorials cover MD data and general datasets like MNIST, making it accessible for users across domains.
9.NeuralTSNE opens the door to future extensions such as reweighted embeddings, spectral maps, and direct use of the learned coordinates in biasing simulations. It fills a key gap in ML tools for physics-informed MD analysis.
10.This implementation democratizes access to parametric t-SNE for chemists and physicists, offering a practical and extensible tool for understanding molecular behavior beyond intuition-driven reaction coordinates.
💻Code: github.com/NeuralTSNE/Neural…
📜Paper: arxiv.org/abs/2505.16476
#MolecularDynamics #DimensionalityReduction #tSNE #NeuralNetworks #CollectiveVariables #UnsupervisedLearning #ComputationalChemistry #NeuralTSNE #EnhancedSampling
5
19
1,283

21 May 2025
What have metal nanoclusters and #proteins in common? Structural transitions that are hard to simulate! #MachineLearning #collectiveVariables enable computing free energy landscape, transition rates, and non-equilibrium behaviour. @AntTinti @IOPscience
👇 iopscience.iop.org/article/1…
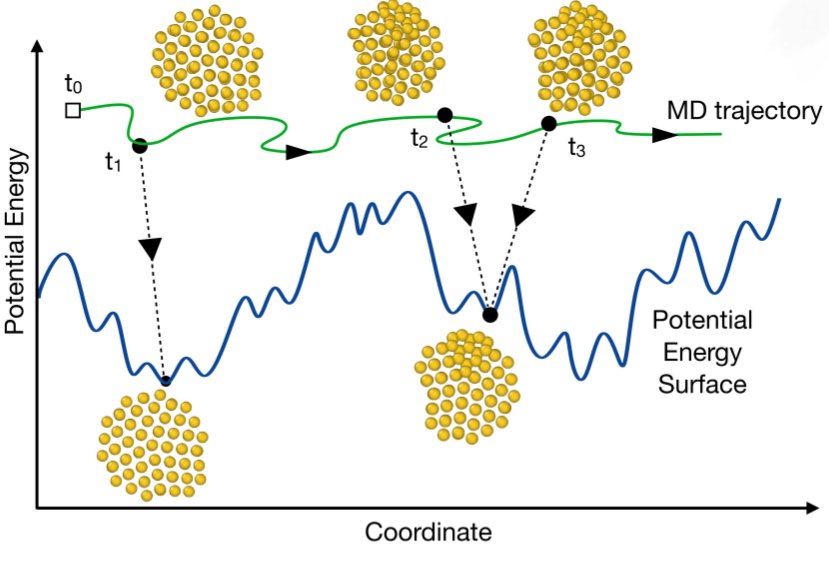
3
13
286
4 Dec 2024
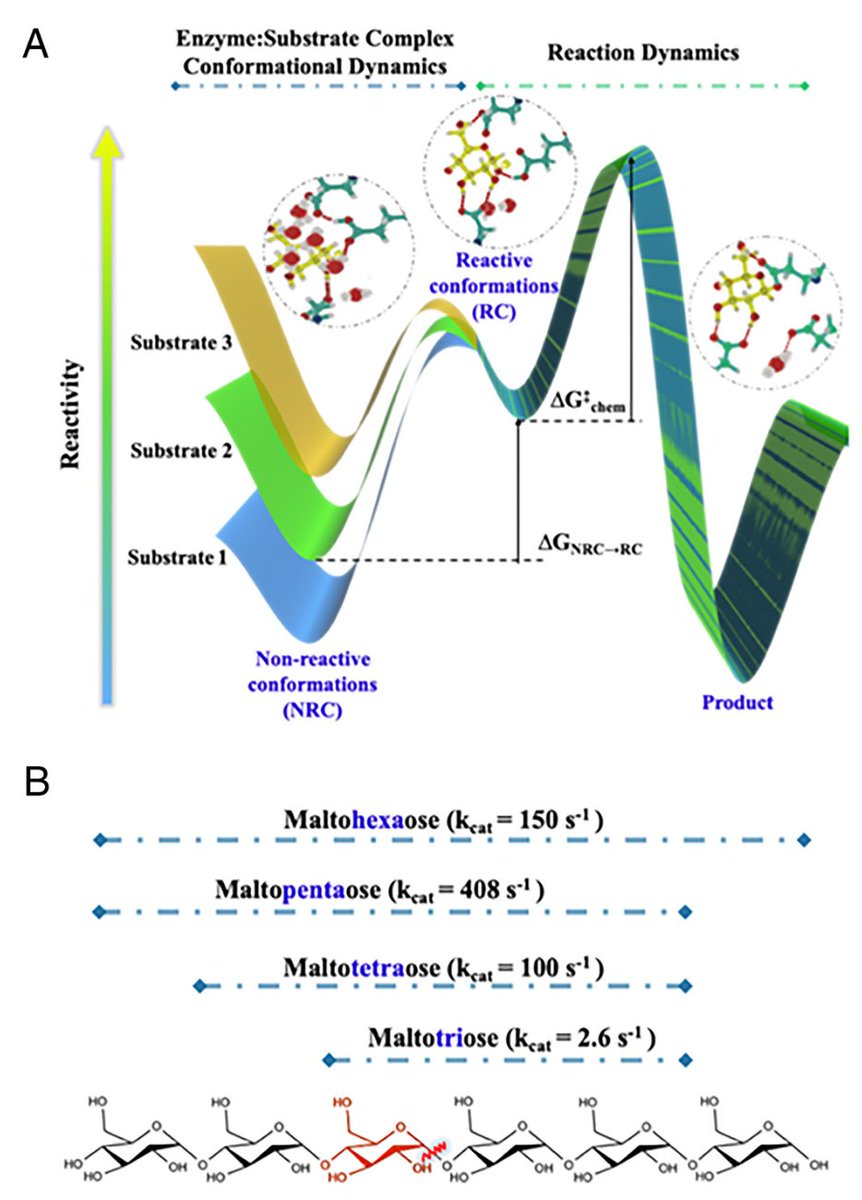
Correlating enzymatic reactivity for different substrates using transferable data-driven collective variables @PNASNews
1. This study introduces machine learning-based collective variables (ML-CVs) for enzymatic catalysis, demonstrating their transferability across substrates with shared binding poses. This innovation streamlines simulations, reducing computational effort and manual intervention.
2. Focusing on human pancreatic α-amylase (HPA), a target for type-II diabetes treatment, the research correlates reactive conformations (RCs) with experimental catalytic activity for four malto-oligosaccharides. The approach captures complex transitions from nonreactive to reactive states.
3. Using Deep Contact and Deep Water CVs, the study identifies three distinct substrate binding modes (NRC1, NRC2, and RC). These CVs accurately describe enzyme-substrate interactions and hydration dynamics in the active site.
4. Remarkably, the ML-CVs trained on maltopentaose were transferable to other substrates, bypassing the need for system-specific CVs. This result validates the generalizability of the approach for similar catalytic systems.
5. Enhanced sampling techniques, such as OPES, reveal detailed free energy surfaces (FES), elucidating the entropic and energetic contributions to enzymatic reactivity. These insights align strongly with experimental catalytic rates (kcat) for all substrates.
6. The findings highlight how transferable ML-CVs can accelerate drug discovery by efficiently evaluating drug efficacy across inhibitor generations. This is particularly relevant for designing derivatives of parent drugs.
7. Future directions include refining ML-CVs for diverse binding modes and scaffolds, broadening their applicability across other enzymatic systems while integrating additional physical and chemical descriptors.
@GroupParrinello @uraucci87 @SudipDa09195612 @IITalk
💻Code: plumed-nest.org/eggs/24/025
📜Paper: pnas.org/doi/10.1073/pnas.24…
#EnzymaticCatalysis #MachineLearning #Bioinformatics #DrugDiscovery #CollectiveVariables #MolecularDynamics
4
32
1,856

31 May 2022
J Chem. Theory Comput.
Exploration vs Convergence Speed in Adaptive-Bias #EnhancedSampling
@inve_michele @GroupParrinello @JCIM_JCTC
#BiasedSampling #moleculardynamics #collectivevariables #PhaseSpace
pubs.acs.org/doi/10.1021/acs…
2
14
30 May 2022
J Chem. Theory Comput.
Learning Efficient, Collective #MonteCarlo Moves with #VariationalAutoencoders
@JCIM_JCTC #autoencoders #collectivevariables #EnhancedSampling #BiasedSampling
pubs.acs.org/doi/10.1021/acs…
1
2

8 Sep 2020
Good morning from CET, @LatinXChem! Ever had troubles defining #collectivevariables? Or had too many to choose from for your #metadynamics run? This thread might have solutions for you ;) #LatinXChemTheo #compchem
7 Sep 2020

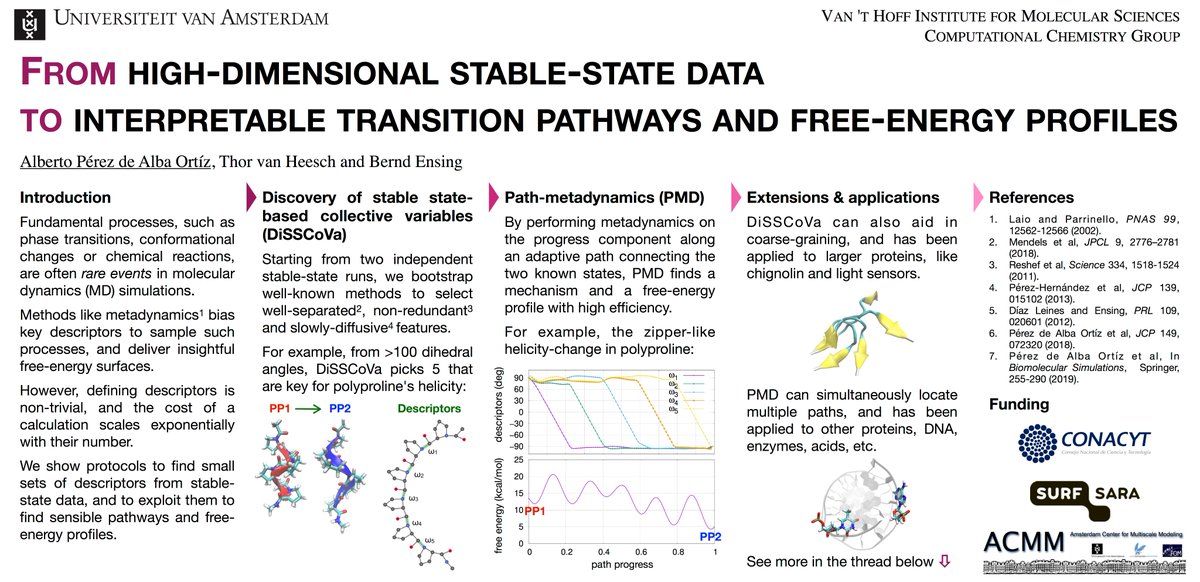
Hi, @LatinXChem! This is my work with @thorvanheesch & @BerndEnsing on data mining & path-based enhanced sampling. Our goal is to go "From high-dimensional stable-state data to interpretable transition pathways and free-energy profiles" #LatinXChem #LatinXChemTheo #Theo154 (1/7).
2