
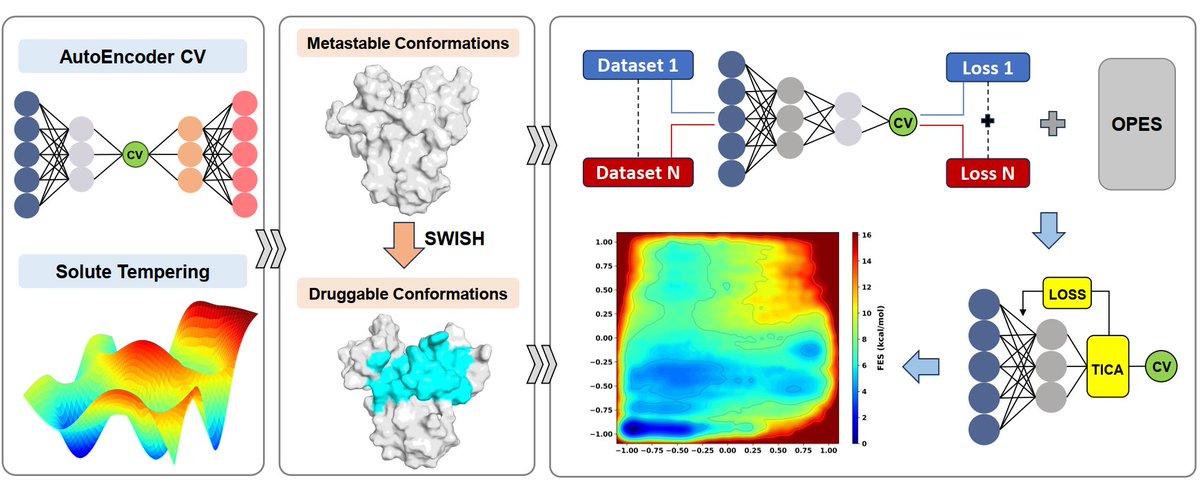
Targeting the intrinsically disordered AR-NTD through a machine learning-based enhanced sampling workflow
In our new work published in @NatureComms , co-first authors Kai Zhu @KaiZhu560347 , Huating Wang, and Jintu Zhang report a computational-experimental strategy for targeting the intrinsically disordered N-terminal domain of the androgen receptor (AR-NTD).
🔬 1. Why AR-NTD?
Most approved androgen receptor antagonists target the ligand-binding domain. However, drug-resistant prostate cancer can bypass this route through AR mutations, amplification, and splice variants. Since these resistance mechanisms often retain the N-terminal domain, AR-NTD represents an attractive but challenging therapeutic target.
🧠 2. What is the key idea?
For intrinsically disordered proteins, the target is not one static structure, but a dynamic ensemble.
By combining enhanced sampling simulations with machine learning-based collective variables, we mapped the conformational landscape of the Tau-5 region of AR-NTD and identified transient druggable conformations.
⚙️ 3. What did we find?
The workflow integrates REST3, AutoEncoder-based collective variables, SWISH, OPES, and Deep-TICA. It identified nine metastable conformations and revealed key features of ligand recognition, including π–π stacking, aromatic residues, and structured water-mediated hydrogen-bond networks.
💡 4. What did this enable?
Guided by these druggable conformations, we performed structure-based virtual screening and discovered K53, a rationally designed AR-NTD antagonist.
K53 directly binds the AR-NTD-AF1 region, suppresses AR transcriptional activity, downregulates canonical AR target genes, and shows potent anti-proliferative activity in enzalutamide-resistant prostate cancer cells. 🎯
🚀 Overall
This work highlights how machine learning-enhanced molecular simulations can help transform dynamic IDP ensembles into actionable opportunities for rational drug discovery, providing a potential roadmap for targeting other challenging intrinsically disordered proteins.
Read the full article here: nature.com/articles/s41467-0…
#DrugDiscovery #AIforScience #MachineLearning #MolecularDynamics #EnhancedSampling #IDP #AndrogenReceptor #ComputationalBiology #ComputationalChemistry #MedicinalChemistry #MolecularSimulation #AI4DrugDiscovery
1
84
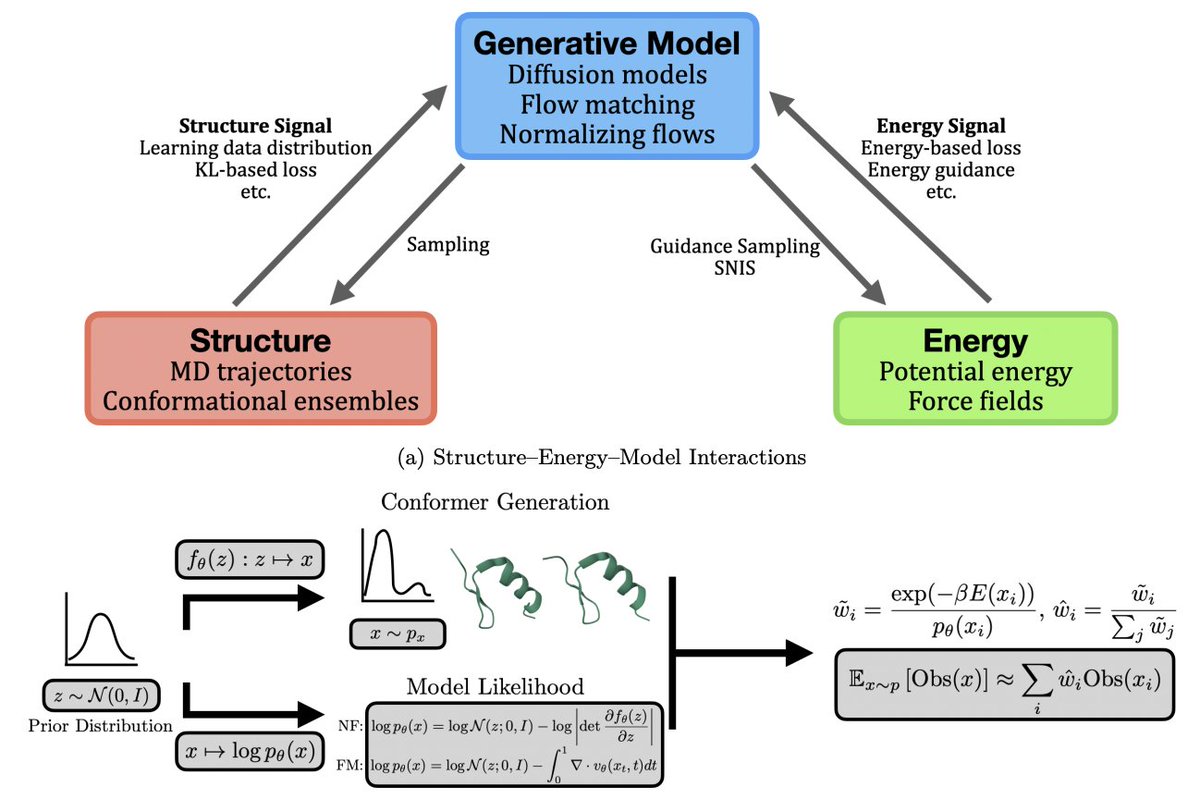
Learning Structure, Energy, and Dynamics: A Survey of Artificial Intelligence for Protein Dynamics
1. The survey maps the protein-dynamics AI landscape into three complementary training signals: learning from structural ensembles/trajectories, learning from physical energy signals (Boltzmann learning), and learning to accelerate or replace components of molecular dynamics (ML force fields, coarse graining, and collective variables).
2. A key theme is the shift from inference-time “make AlphaFold diverse” heuristics to explicit generative modeling of p(x|sequence) for equilibrium ensembles, using diffusion, flow matching, and latent language-modeling approaches that can be trained end-to-end on ensemble data and sampled efficiently.
3. For equilibrium ensemble generation, it highlights how modern methods incorporate stronger priors and broader conditioning: MSA-free sequence-conditioned generators (e.g., latent diffusion with protein language models), temperature/thermodynamic conditioning, and energy-conditioned sampling to target distinct conformational states rather than a single dominant fold.
4. It emphasizes practical failure modes in purely data-driven ensemble learning—limited physical realism, scarcity of diverse dynamic data, and PDB conformational bias—and surveys mitigation strategies such as force/energy guidance, physics-informed objectives (e.g., Fokker–Planck supervision), experimental stability signals, and dataset reweighting via structural clustering.
5. The review extends “dynamics generation” beyond i.i.d. conformers to explicit trajectory models p(X|x1): (i) learned long-timestep transition kernels (MCMC-style), (ii) autoregressive frame prediction with improved temporal scalability (including state-space-model adaptations), and (iii) one-shot “trajectory-as-video” generation that outputs full time-ordered coordinate sequences in a single pass.
6. A central innovation thread is energy-driven learning: Boltzmann generators and related samplers that use energies/forces to learn proposals for the Boltzmann distribution, then correct residual bias with self-normalized importance sampling or annealing/SMC-style procedures, with effective sample size (ESS) used as a key reliability diagnostic.
7. The survey contrasts exact-likelihood normalizing-flow Boltzmann generators (enabling principled reweighting) with likelihood-free diffusion/flow approaches trained from energetic supervision, and discusses the core tradeoff: thermodynamic guarantees vs scalability, symmetry handling, and computational cost of likelihood/energy evaluations.
8. It also covers “physics-aware adaptation” of pretrained protein generators: post-training alignment and inference-time steering that tilt a base generator toward lower-free-energy or constraint-satisfying samples using energies, forces, projections, or CV/observable-based biases—aiming to retrofit thermodynamic meaning without retraining from scratch.
9. On the simulation side, it reviews how ML accelerates or upgrades MD via: (i) machine-learning potentials approaching QM fidelity while scaling to large biomolecular systems (often via fragmentation, Δ-learning, and explicit long-range terms), (ii) learned coarse-grained models that approximate potentials of mean force for longer timescales, and (iii) ML-derived collective variables for enhanced sampling using dimensionality reduction, kinetic learning, RL/adaptive sampling, and differentiable generative constraints.
10. The survey closes by curating key datasets (static PDB and distilled AFDB/AFESM; MD trajectory corpora like ATLAS, DynamicPDB, mdCATH, MISATO, DD-13M; and IDP/experimental resources like BMRB and SASBDB) and framing open challenges: scalability, thermodynamic consistency, kinetic fidelity, dataset/force-field bias, and principled integration with experimental constraints.
📜Paper: arxiv.org/abs/2604.25244
#ProteinDynamics #ComputationalBiology #MolecularDynamics #GenerativeAI #DiffusionModels #FlowMatching #BoltzmannGenerators #MachineLearningPotentials #CoarseGraining #EnhancedSampling
1
4
40
2,438
Fast sampling of protein conformational dynamics @ScienceAdvances
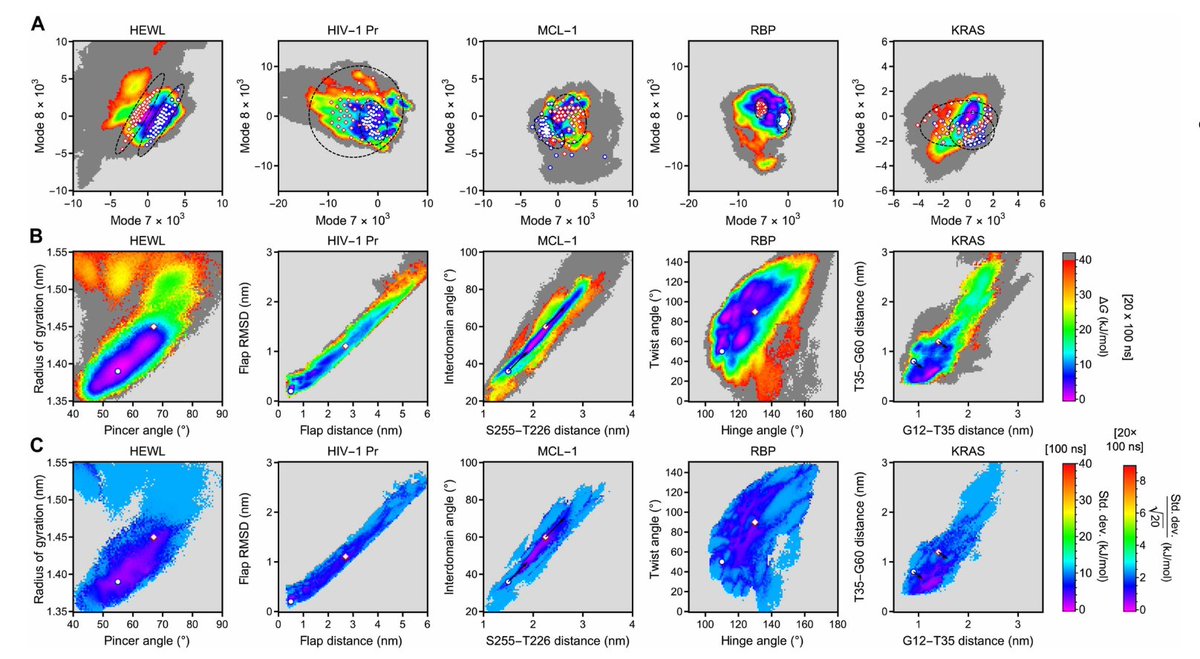
1. Sauer et al. show that the key collective variables (CVs) needed to drive enhanced sampling of protein conformational transitions are encoded in anharmonic low-frequency vibrations, and these CVs can be extracted from short unbiased MD without any prior knowledge of the transition.
2. Core idea: use FRESEAN (frequency-selective anharmonic mode analysis) at (near) zero frequency to isolate collective motions with minimal restoring forces—i.e., “paths of least resistance” for conformational change—avoiding the limitations of harmonic/quasiharmonic normal modes in the low-frequency, diffusive regime.
3. Practical pipeline: run 20 ns unbiased all-atom MD, align trajectories, coarse-grain to a 2-bead-per-residue representation (1 for Gly), compute velocity time-correlation matrices, Fourier transform to frequency domain, then take eigenvectors at zero frequency. Modes 1–6 correspond to translation/rotation and are discarded; modes 7 capture internal anharmonic low-frequency vibrations.
4. Reproducibility is a central result: across 5 independent 20 ns replicas per protein, the low-frequency modes (especially the 2D subspace spanned by modes 7–8) are consistently recovered, unlike PCA/quasiharmonic modes whose replica-to-replica agreement remains poor even with much longer trajectories.
5. Enhanced sampling step: use modes 7 and 8 as CVs in well-tempered metadynamics (100 ns per run; reported as <24 hours on a single GPU). Across 5 proteins × 5 replicas, 22/25 runs (88%) sample known “closed↔open” transitions within 100 ns; extending to 160 ns yields full sampling for all replicas.
6. Benchmark set spans diverse challenges: HEWL (disulfide-stabilized), HIV-1 protease (homodimer), MCL-1 (allosteric/druggable dynamics), ribose-binding protein (multi-domain hinge motion), and GDP-bound KRAS (switch-region dynamics). The same FRESEAN-to-metadynamics protocol is applied across all systems.
7. Free-energy landscapes (FES) become both fast and statistically controlled by running 20 parallel metadynamics replicas (20 × 100 ns) using the same FRESEAN CVs: single-run uncertainties are typically < ±10 kJ/mol, and averaging reduces standard error to < ±3 kJ/mol, enabling reproducible thermodynamic ensembles rather than just qualitative transitions.
8. Comparison to “hand-crafted” geometric CVs from prior literature is informative: biasing along FRESEAN modes often follows lower-free-energy transition routes and tends to keep sampling within the native folded ensemble, whereas geometric CVs can push systems into partially unfolded high-entropy states (most notably KRAS when biased by residue–residue distances).
9. The authors quantify cross-CV reweighting fidelity using Shannon entropy and Bhattacharyya coefficients: on average, ensembles generated by biasing along low-frequency vibrational CVs preserve at least as much (often more) information when reweighted into geometric-variable space than the reverse, supporting the claim that these vibrations are broadly suitable, system-agnostic CVs.
10. Implication for computational biology/ML: the method enables high-throughput generation of conformational ensembles and FESs (including mutants/conditions), helping address the dataset bottleneck for next-generation sequence→structure→dynamics models beyond single static folds or single thermodynamic states.
💻Code: github.com/HeydenLabASU-coll…
📜Paper: doi.org/10.1126/sciadv.aea46…
#MolecularDynamics #EnhancedSampling #Metadynamics #ProteinDynamics #FreeEnergy #ComputationalBiophysics #CollectiveVariables #FRESEAN #GROMACS #PLUMED
16
70
4,180

24 Dec 2025
🎄 Xmas Eve present for catalysis nerds 🎁
New paper out in J. Catal.: protonation dynamics of ethanol–water clusters in H-ZSM-5, using ML potentials metadynamics.
Solvation, protons, free-energies...
👉 sciencedirect.com/science/ar…
#ML #enhancedsampling keeps delivering 🚀
1
3
122

14 Nov 2025
Our team @snbkol introduces PathGennie (just published in @JCIM_JCTC), a fast and unbiased computational framework for uncovering rare molecular events such as drug unbinding and protein folding.
Instead of using any biased forces or elevated temperatures, PathGennie launches ultrashort unbiased MD trajectories and selectively propagates only those that show real progress toward the transition. This lets us generate complete rare-event pathways in just 10–100 ps, while preserving true molecular dynamics.
Full credit goes to my PhD student Dibyendu Maity @MAITYDIBYENDU99 for formulating and implementing the PathGennie code. Thanks to @ANRFIndia for funding.
✨ Key Highlights:
• Identified 7 distinct unbinding routes for benzene from T4 lysozyme
• Recovered all major exit pathways for imatinib from Abl kinase within a handful of attempts
• Captured folding/unfolding pathways of Trp-cage and Protein G that align with minimum free-energy paths
• Weighted Ensemble (WE) simulations converge 3–4× faster when initialised with PathGennie-generated pathways
• Path finding works even with suboptimal CVs
🔗 Try our code: github.com/TeamSuman/PathGen…
📄 Read the paper: pubs.acs.org/doi/10.1021/acs…
#CompChem #RareEvent #EnhancedSampling #MolecularDynamics #PathSampling #DrugDiscovery
14
62
481
54,027

15 Oct 2025
Enhancing Sampling for Efficient Learning of Coarse-Grained Machine Learning Potentials
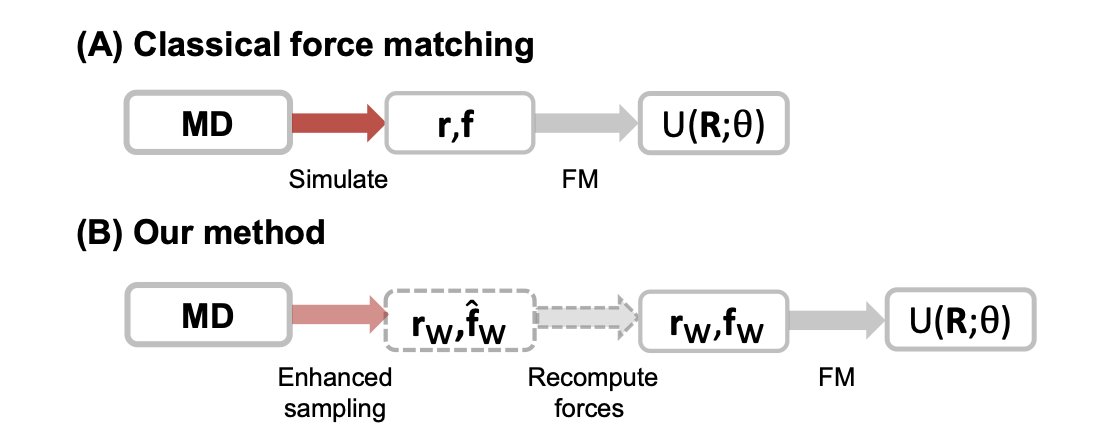
1. A new method for improving the accuracy and efficiency of coarse-grained molecular dynamics simulations has been proposed by Weilong Chen and colleagues. The study introduces an enhanced sampling strategy to address the limitations of current coarse-grained machine learning potentials (MLPs), which often struggle with insufficient sampling in transition regions and require long atomistic trajectories to achieve convergence.
2. The core innovation lies in the use of biased sampling along coarse-grained degrees of freedom to generate training data. By applying a bias potential and then recomputing forces with respect to the unbiased potential, the method can produce equilibrated data more quickly and enrich sampling in transition regions, all while preserving the correct potential of mean force (PMF).
3. The effectiveness of this approach is demonstrated through simulations of the Müller–Brown potential and capped alanine. The results show significant improvements in data efficiency and model accuracy compared to traditional force matching methods that rely on unbiased equilibrium data. Models trained on biased datasets required fewer samples to achieve lower error rates and better coverage of transition states.
4. The study employs popular enhanced sampling techniques such as umbrella sampling and well-tempered metadynamics. These methods are shown to accelerate the exploration of configuration space, providing more uniform coverage of both energy basins and transition regions. The use of a graph neural network potential without prior physics-based terms further enhances the model's ability to learn complex interactions directly from data.
5. The findings suggest that enhanced sampling can serve as a powerful and general framework for constructing accurate and data-efficient coarse-grained MLPs. This approach not only improves the accuracy of force matching but also reduces the computational cost associated with generating training data, making it a promising direction for future research in coarse-grained molecular modeling.
📜Paper: arxiv.org/abs/2510.11148v1
#MolecularDynamics #CoarseGraining #MachineLearning #EnhancedSampling #ComputationalBiology
1
7
870

22 Jun 2025
🚀 Based on Voronoi CV-based OPES DPLRMD, our new study reveals how electric fields & air-water interfaces switch glycine tautomerism mechanisms @JCIM_JCTC.
🔗 Article: doi.org/10.1021/acs.jctc.5c0…
📂 Data: github.com/Zhang-pchao/resea…
#compchem #machinelearning #enhancedsampling
5
1
9
1,035
23 May 2025
NeuralTSNE: A Python Package for the Dimensionality Reduction of Molecular Dynamics Data Using Neural Networks
1.NeuralTSNE is a new Python package implementing parametric t-SNE using neural networks for dimensionality reduction of molecular dynamics (MD) data. It enables high-fidelity mapping of complex molecular trajectories into low-dimensional representations ideal for analysis and visualization.
2.Unlike standard t-SNE, NeuralTSNE uses a neural network to learn a continuous mapping from high-dimensional feature space to low-dimensional latent space. This allows reusability of the trained model on new data—a key advantage for MD workflows.
3.The algorithm minimizes the Kullback-Leibler divergence between high-dimensional and reduced-space distance distributions using a feedforward neural network trained via PyTorch and PyTorch Lightning. It supports GPU acceleration and is easy to integrate into modern ML pipelines.
4.NeuralTSNE is designed for usability: it can be run as a command-line tool or imported as a Python module. Its API is consistent with scikit-learn’s interface, providing familiar fit and predict functions.
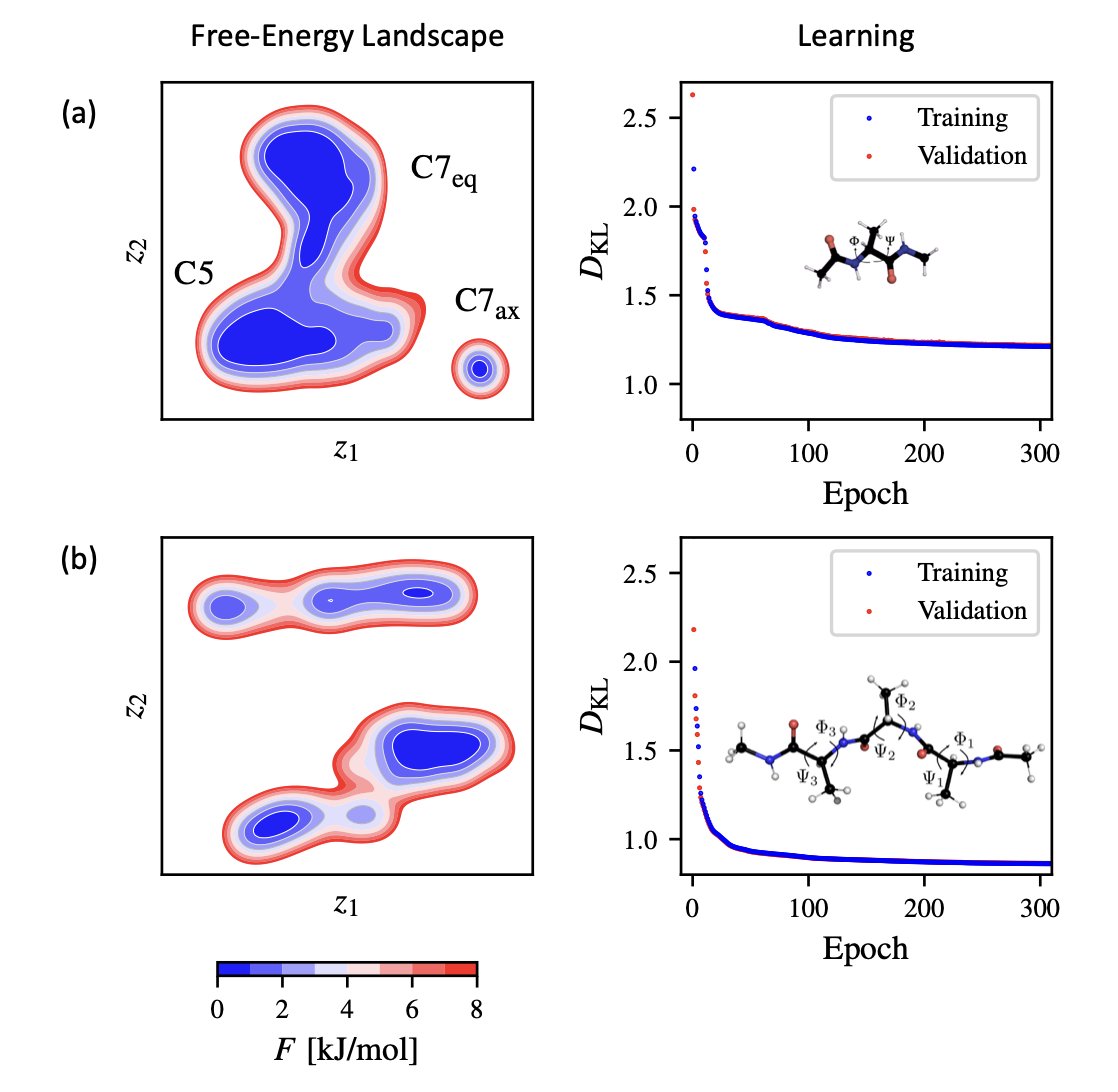
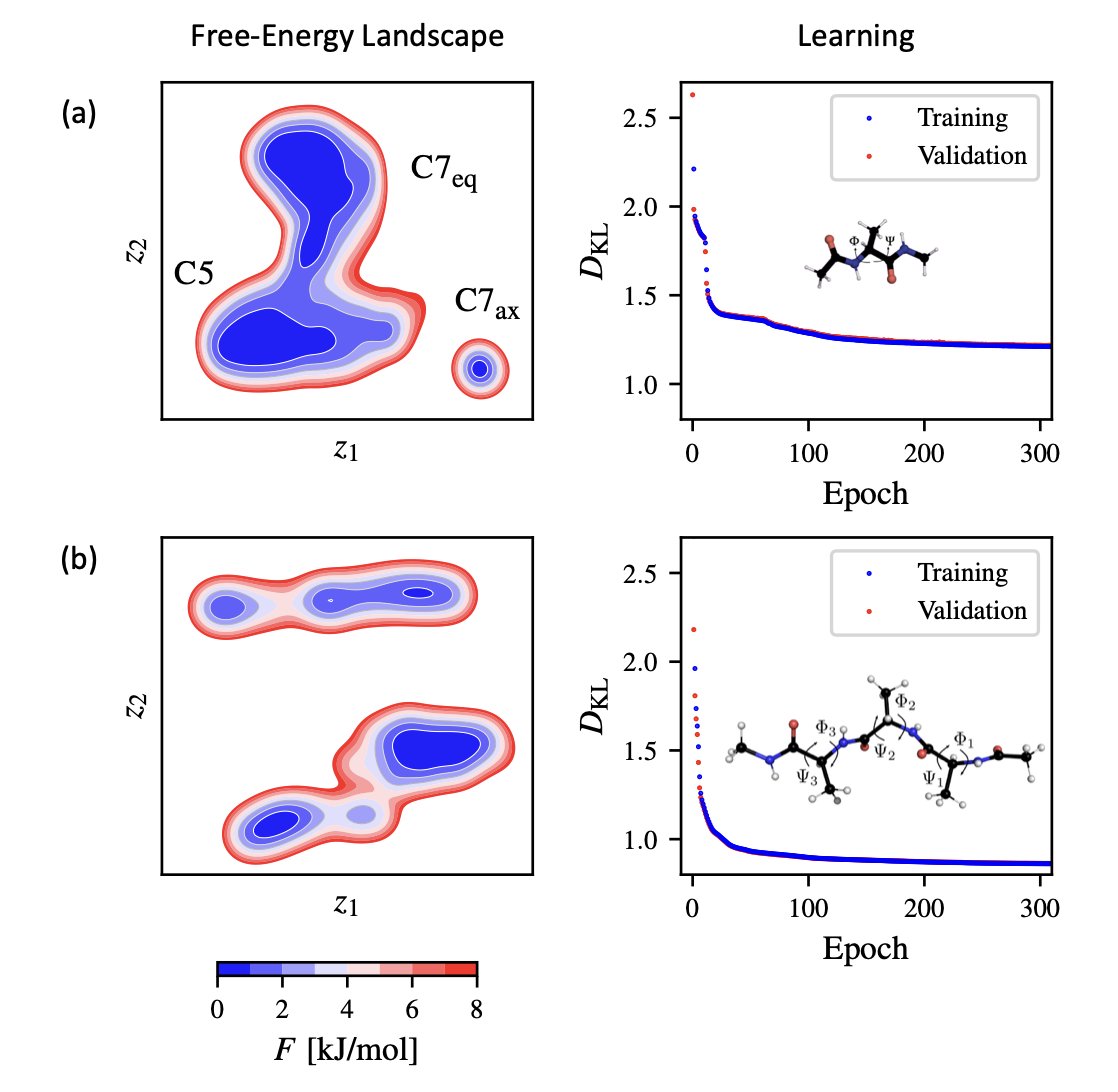
5.The package is demonstrated on MD datasets from alanine dipeptide and tetrapeptide simulations. It successfully reconstructs known free-energy landscapes, recovering metastable states and transition barriers with remarkable precision.
6.In the alanine dipeptide case, NeuralTSNE identifies three key states—C7eq, C5, and C7ax—matching established dihedral-based mappings. For the tetrapeptide, the model identifies complex multi-state landscapes, highlighting its capacity to handle high-dimensional, rugged data.
7.Unlike static t-SNE implementations, NeuralTSNE generalizes well to out-of-sample data, making it suitable for online analysis and enhanced sampling simulations. The authors plan to integrate it with PLUMED to guide MD trajectories using learned coordinates.
8.The package provides full reproducibility: examples, testing frameworks, CI/CD pipelines, and documentation are available on GitHub. Tutorials cover MD data and general datasets like MNIST, making it accessible for users across domains.
9.NeuralTSNE opens the door to future extensions such as reweighted embeddings, spectral maps, and direct use of the learned coordinates in biasing simulations. It fills a key gap in ML tools for physics-informed MD analysis.
10.This implementation democratizes access to parametric t-SNE for chemists and physicists, offering a practical and extensible tool for understanding molecular behavior beyond intuition-driven reaction coordinates.
💻Code: github.com/NeuralTSNE/Neural… 📜Paper: arxiv.org/abs/2505.16476
#MolecularDynamics #DimensionalityReduction #tSNE #NeuralNetworks #CollectiveVariables #UnsupervisedLearning #ComputationalChemistry #NeuralTSNE #EnhancedSampling
3
31
1,849
23 May 2025
NeuralTSNE: A Python Package for the Dimensionality Reduction of Molecular Dynamics Data Using Neural Networks
1.NeuralTSNE is a new Python package implementing parametric t-SNE using neural networks for dimensionality reduction of molecular dynamics (MD) data. It enables high-fidelity mapping of complex molecular trajectories into low-dimensional representations ideal for analysis and visualization.
2.Unlike standard t-SNE, NeuralTSNE uses a neural network to learn a continuous mapping from high-dimensional feature space to low-dimensional latent space. This allows reusability of the trained model on new data—a key advantage for MD workflows.
3.The algorithm minimizes the Kullback-Leibler divergence between high-dimensional and reduced-space distance distributions using a feedforward neural network trained via PyTorch and PyTorch Lightning. It supports GPU acceleration and is easy to integrate into modern ML pipelines.
4.NeuralTSNE is designed for usability: it can be run as a command-line tool or imported as a Python module. Its API is consistent with scikit-learn’s interface, providing familiar fit and predict functions.
5.The package is demonstrated on MD datasets from alanine dipeptide and tetrapeptide simulations. It successfully reconstructs known free-energy landscapes, recovering metastable states and transition barriers with remarkable precision.
6.In the alanine dipeptide case, NeuralTSNE identifies three key states—C7eq, C5, and C7ax—matching established dihedral-based mappings. For the tetrapeptide, the model identifies complex multi-state landscapes, highlighting its capacity to handle high-dimensional, rugged data.
7.Unlike static t-SNE implementations, NeuralTSNE generalizes well to out-of-sample data, making it suitable for online analysis and enhanced sampling simulations. The authors plan to integrate it with PLUMED to guide MD trajectories using learned coordinates.
8.The package provides full reproducibility: examples, testing frameworks, CI/CD pipelines, and documentation are available on GitHub. Tutorials cover MD data and general datasets like MNIST, making it accessible for users across domains.
9.NeuralTSNE opens the door to future extensions such as reweighted embeddings, spectral maps, and direct use of the learned coordinates in biasing simulations. It fills a key gap in ML tools for physics-informed MD analysis.
10.This implementation democratizes access to parametric t-SNE for chemists and physicists, offering a practical and extensible tool for understanding molecular behavior beyond intuition-driven reaction coordinates.
💻Code: github.com/NeuralTSNE/Neural…
📜Paper: arxiv.org/abs/2505.16476
#MolecularDynamics #DimensionalityReduction #tSNE #NeuralNetworks #CollectiveVariables #UnsupervisedLearning #ComputationalChemistry #NeuralTSNE #EnhancedSampling
5
19
1,283
16 May 2025
Hamiltonian replica exchange augmented with diffusion-based generative models and importance sampling to assess biomolecular conformational basins and barriers
1.This study introduces a hybrid framework that enhances Hamiltonian replica exchange (REST2) simulations with denoising diffusion probabilistic models (DDPMs) and importance sampling to more efficiently map biomolecular free-energy landscapes.
2.By adapting DDPMs to REST2 instead of traditional temperature-based replica exchange (TREM), the method significantly reduces the number of required replicas while maintaining accurate sampling—scaling better with system size and computational cost.
3.A key innovation is treating rescaled potential energy as a fluctuating variable in the DDPM framework, allowing the model to learn joint distributions over structure and Hamiltonian perturbation, improving resolution in high-barrier regions.
4.Benchmarking on the mini-protein CLN025 shows that DDPM-augmented REST2 achieves free-energy landscape quality comparable to DDPM-TREM, but with 2.7× fewer replicas and higher exchange rates, demonstrating its sampling efficiency.
5.In both REST2 and TREM, raw simulations struggle to resolve transition states; DDPM refines the undersampled regions and uncovers full transition pathways between metastable basins without needing additional simulation time.
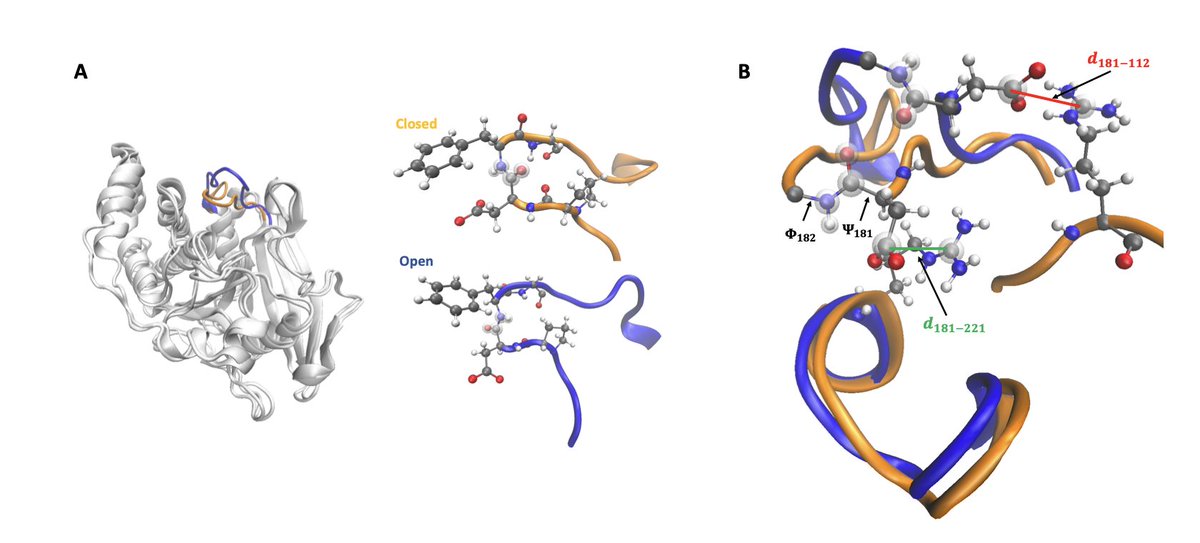
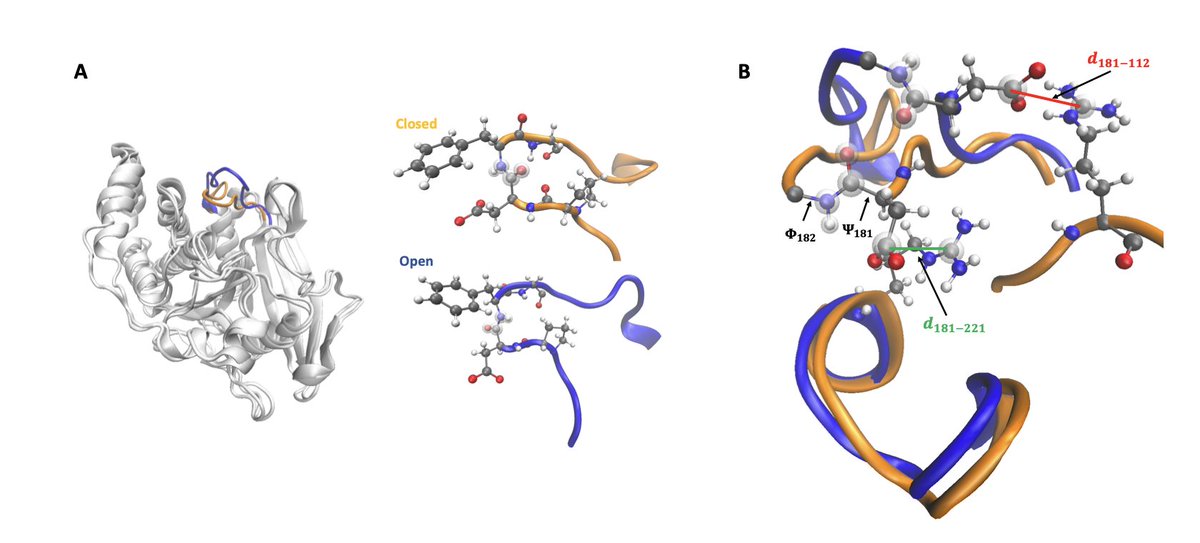
6.For the larger enzyme PTP1B, the framework successfully reveals the loop opening/closing transition in the active site WPD loop, recovering a 12 kcal/mol free-energy barrier consistent with prior biased simulations and experimental data.
7.To further improve sampling in rare-event regions, the authors introduce an iterative biasing scheme where DDPM-generated high-temperature statistics are used to bias REST2 simulations along known slow degrees of freedom, such as torsion angles.
8.This approach not only improves the sampling of the transition path but also corrects misestimated free-energy differences between states, yielding more accurate thermodynamic and mechanistic interpretations.
9.The model detects multiple viable transition routes and evaluates their likelihoods using committor analysis, providing a physically grounded way to validate generative sampling predictions in molecular systems.
10.DDPM alone cannot overcome poor sampling; exchanges between replicas are essential for capturing folded–unfolded transitions. Without exchanges, generative models trained on incomplete data fail to predict missing conformations.
11.Compared to umbrella sampling, the combined DDPM REST2 importance sampling strategy matches the benchmark 1D free-energy profiles in relevant regions, and outperforms it in capturing complex, multidimensional barriers.
12.This study extends the role of generative models from post hoc denoising to active participants in sampling workflows, providing a principled way to accelerate convergence and extract mechanistic insights from high-dimensional MD data.
📜Paper: arxiv.org/abs/2505.08357
#EnhancedSampling #DiffusionModels #ReplicaExchange #ProteinDynamics #FreeEnergyLandscapes #ComputationalBiophysics #MolecularSimulation #AI4Science #REST2 #BiomolecularMechanisms
3
645
16 May 2025
Hamiltonian replica exchange augmented with diffusion-based generative models and importance sampling to assess biomolecular conformational basins and barriers
1.This study introduces a hybrid framework that enhances Hamiltonian replica exchange (REST2) simulations with denoising diffusion probabilistic models (DDPMs) and importance sampling to more efficiently map biomolecular free-energy landscapes.
2.By adapting DDPMs to REST2 instead of traditional temperature-based replica exchange (TREM), the method significantly reduces the number of required replicas while maintaining accurate sampling—scaling better with system size and computational cost.
3.A key innovation is treating rescaled potential energy as a fluctuating variable in the DDPM framework, allowing the model to learn joint distributions over structure and Hamiltonian perturbation, improving resolution in high-barrier regions.
4.Benchmarking on the mini-protein CLN025 shows that DDPM-augmented REST2 achieves free-energy landscape quality comparable to DDPM-TREM, but with 2.7× fewer replicas and higher exchange rates, demonstrating its sampling efficiency.
5.In both REST2 and TREM, raw simulations struggle to resolve transition states; DDPM refines the undersampled regions and uncovers full transition pathways between metastable basins without needing additional simulation time.
6.For the larger enzyme PTP1B, the framework successfully reveals the loop opening/closing transition in the active site WPD loop, recovering a 12 kcal/mol free-energy barrier consistent with prior biased simulations and experimental data.
7.To further improve sampling in rare-event regions, the authors introduce an iterative biasing scheme where DDPM-generated high-temperature statistics are used to bias REST2 simulations along known slow degrees of freedom, such as torsion angles.
8.This approach not only improves the sampling of the transition path but also corrects misestimated free-energy differences between states, yielding more accurate thermodynamic and mechanistic interpretations.
9.The model detects multiple viable transition routes and evaluates their likelihoods using committor analysis, providing a physically grounded way to validate generative sampling predictions in molecular systems.
10.DDPM alone cannot overcome poor sampling; exchanges between replicas are essential for capturing folded–unfolded transitions. Without exchanges, generative models trained on incomplete data fail to predict missing conformations.
11.Compared to umbrella sampling, the combined DDPM REST2 importance sampling strategy matches the benchmark 1D free-energy profiles in relevant regions, and outperforms it in capturing complex, multidimensional barriers.
12.This study extends the role of generative models from post hoc denoising to active participants in sampling workflows, providing a principled way to accelerate convergence and extract mechanistic insights from high-dimensional MD data.
📜Paper: arxiv.org/abs/2505.08357
#EnhancedSampling #DiffusionModels #ReplicaExchange #ProteinDynamics #FreeEnergyLandscapes #ComputationalBiophysics #MolecularSimulation #AI4Science #REST2 #BiomolecularMechanisms
4
22
1,153
27 Apr 2025
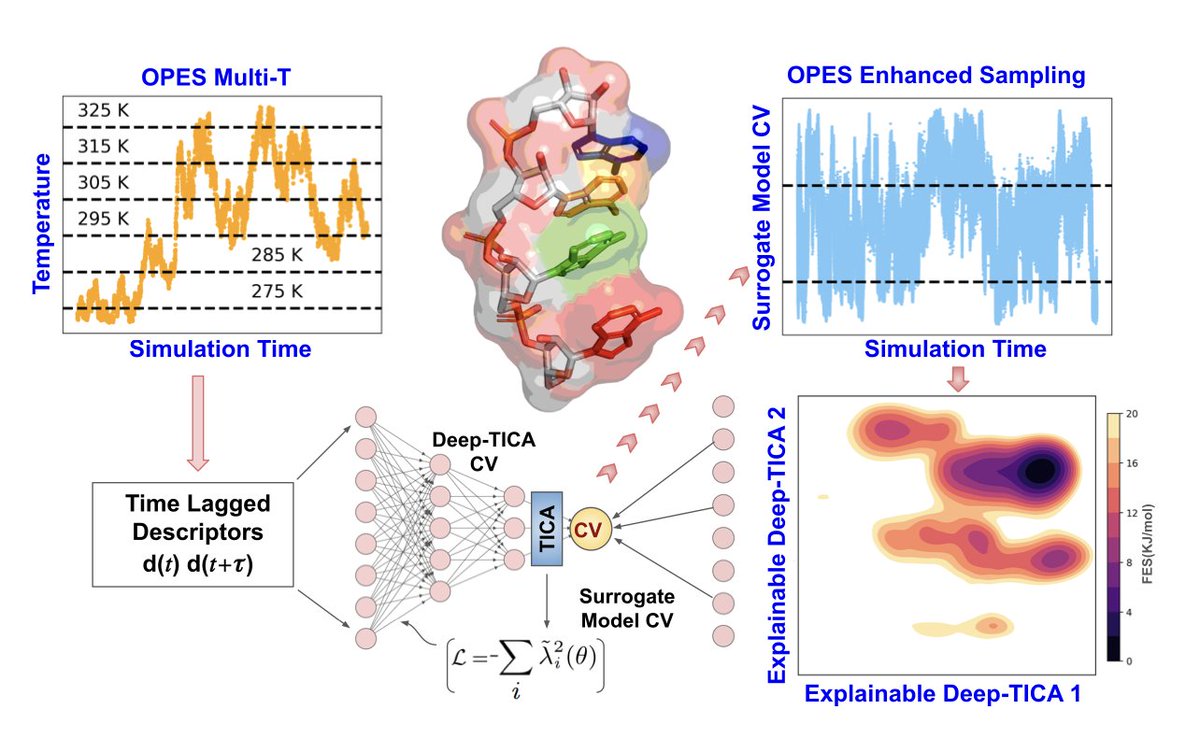
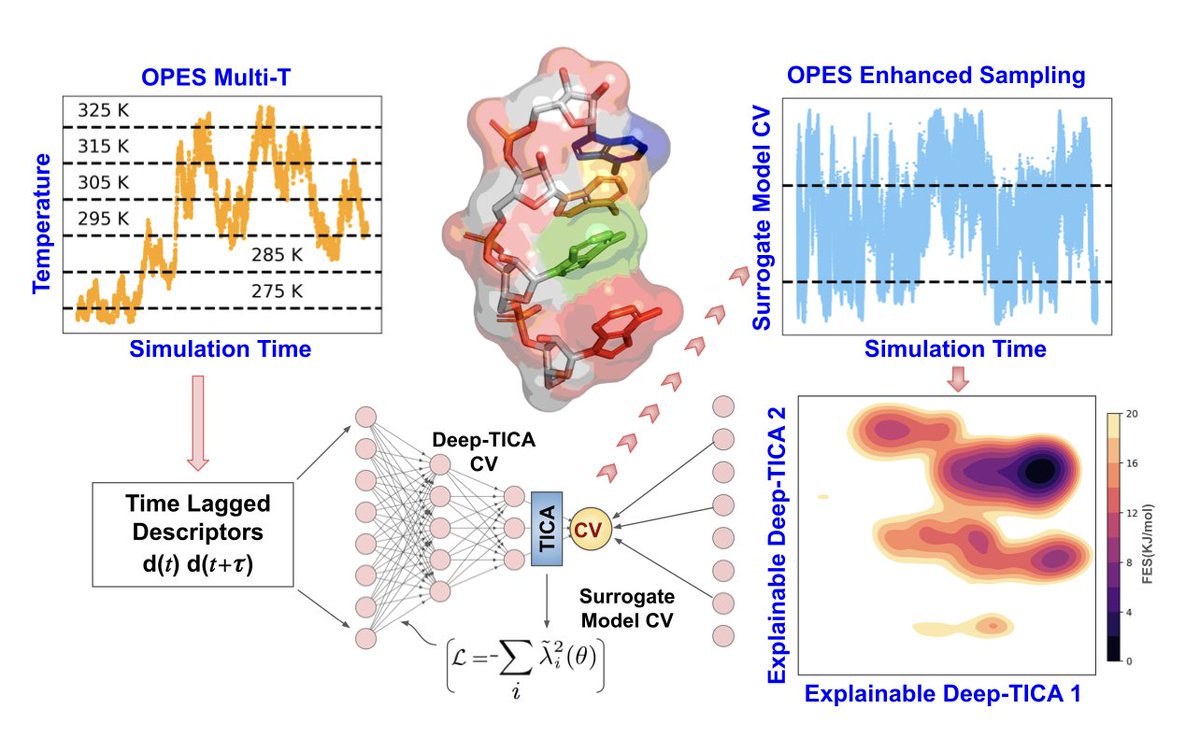
Characterizing RNA Tetramer Conformational Landscape Using Explainable Machine Learning
1. This study introduces an advanced simulation framework that combines explainable machine learning (XAI) with the On-the-Fly Probability Enhanced Sampling (OPES) algorithm to effectively explore the free energy landscapes of RNA tetramers, dramatically improving sampling efficiency.
2. By utilizing XAI, the method distinguishes conformational states, including stacked, intercalated, and random coil structures, with significantly reduced computational cost—achieving two orders of magnitude faster convergence compared to conventional molecular dynamics.
3. The approach employs explainable deep learning models to identify key torsion angles in RNA tetramers that influence slow conformational transitions, offering a mechanistic understanding of RNA dynamics that is often hidden in black-box machine learning models.
4. The framework incorporates a Deep-TICA algorithm, which identifies the slowest degrees of freedom in RNA tetramers, providing crucial insights into the complex free energy landscapes of systems that exhibit substantial structural heterogeneity.
5. The study focuses on RNA tetramers, specifically the AAAA, CCCC, and UUUU sequences, demonstrating how machine learning-guided enhanced sampling can capture complex metastable states that are hard to resolve with traditional methods.
6. One major innovation is the use of surrogate models for collective variables (CVs), which reduce the computational burden by transforming the outputs of deep neural networks into interpretable linear combinations of key molecular descriptors, such as backbone and glycosidic torsion angles.
7. The methodology not only accelerates simulations but also provides interpretable results, such as identifying the specific torsion angles that govern metastable transitions, which could lead to improvements in RNA force fields and a better understanding of RNA conformational dynamics.
8. This work presents a significant advancement in simulating RNA conformational landscapes, offering a framework that can be applied to other biomolecules, including disordered proteins, that exhibit rugged free energy landscapes.
📜Paper: doi.org/10.26434/chemrxiv-20…
#RNA #MachineLearning #Bioinformatics #EnhancedSampling #ComputationalBiology #DeepLearning #MolecularDynamics #XAI #BioMolecularSimulation
1
3
1,004
27 Apr 2025
Characterizing RNA Tetramer Conformational Landscape Using Explainable Machine Learning
1. This study introduces an advanced simulation framework that combines explainable machine learning (XAI) with the On-the-Fly Probability Enhanced Sampling (OPES) algorithm to effectively explore the free energy landscapes of RNA tetramers, dramatically improving sampling efficiency.
2. By utilizing XAI, the method distinguishes conformational states, including stacked, intercalated, and random coil structures, with significantly reduced computational cost—achieving two orders of magnitude faster convergence compared to conventional molecular dynamics.
3. The approach employs explainable deep learning models to identify key torsion angles in RNA tetramers that influence slow conformational transitions, offering a mechanistic understanding of RNA dynamics that is often hidden in black-box machine learning models.
4. The framework incorporates a Deep-TICA algorithm, which identifies the slowest degrees of freedom in RNA tetramers, providing crucial insights into the complex free energy landscapes of systems that exhibit substantial structural heterogeneity.
5. The study focuses on RNA tetramers, specifically the AAAA, CCCC, and UUUU sequences, demonstrating how machine learning-guided enhanced sampling can capture complex metastable states that are hard to resolve with traditional methods.
6. One major innovation is the use of surrogate models for collective variables (CVs), which reduce the computational burden by transforming the outputs of deep neural networks into interpretable linear combinations of key molecular descriptors, such as backbone and glycosidic torsion angles.
7. The methodology not only accelerates simulations but also provides interpretable results, such as identifying the specific torsion angles that govern metastable transitions, which could lead to improvements in RNA force fields and a better understanding of RNA conformational dynamics.
8. This work presents a significant advancement in simulating RNA conformational landscapes, offering a framework that can be applied to other biomolecules, including disordered proteins, that exhibit rugged free energy landscapes.
📜Paper: doi.org/10.26434/chemrxiv-20…
#RNA #MachineLearning #Bioinformatics #EnhancedSampling #ComputationalBiology #DeepLearning #MolecularDynamics #XAI #BioMolecularSimulation
5
15
1,462

23 Mar 2025
Thrilled to join #ACSSpring2025 and meet my friends! Don't miss my talk on how #AI & #EnhancedSampling can describe gene editing & refine #cryoEM maps. We’ll be presenting our latest research on #CRISPR-associated transposon systems other exciting advancements! #compchem #RNA
4
9
96
6,293

This summer we used #moleculardynamics simulations and #enhancedsampling to predict the solvation shell of a ribozyme! Results will be announced at the coming #CASP16 conference, to which we will attend to present our approach. Finger crossed 🤞
13 Nov 2024

CASP16 Abstract Book is online!
predictioncenter.org/casp16/…
Dive into the latest advancements with the #CASP16 Abstract Book, showcasing groundbreaking methods from deep learning, AI-driven model refinement, RNA 3D structure predictions, and ligand docking strategies.
From AlphaFold innovations to unique machine learning approaches, this collection represents the cutting edge in structural biology!

1
13
728
14 Nov 2024
Fast Sampling of Protein Conformational Dynamics
• This study introduces a new approach using anharmonic low-frequency vibrations as collective variables (CVs) for enhanced sampling of protein conformational dynamics, enabling faster and more consistent exploration of protein conformations.
• FRESEAN mode analysis isolates low-frequency vibrations without relying on invalid harmonic assumptions, allowing efficient sampling of dynamic conformational changes. This analysis can be performed on nanosecond simulations, making it accessible and reproducible.
• Applied to five diverse proteins, FRESEAN identified reproducible vibrational modes that served as effective CVs in metadynamics, capturing conformational transitions with minimal computational cost.
• Enhanced sampling simulations using these CVs achieved up to six-fold increased sampling of dynamic range compared to unbiased simulations, offering fast, high-throughput sampling critical for training datasets in future protein dynamics prediction models.
• Results demonstrate the capability to generate free energy surfaces from sampled data, revealing both known and novel conformational states, with high reproducibility across multiple replicas.
• This efficient sampling method opens pathways for comprehensive protein dynamics datasets, aiding the integration of dynamic conformational ensembles into predictive machine learning models.
@sthitadhi_m
📜Paper: arxiv.org/abs/2411.08154
#ProteinDynamics #Bioinformatics #MolecularSimulations #EnhancedSampling #FRESEAN #Metadynamics

2
11
46
3,130
7 Nov 2024
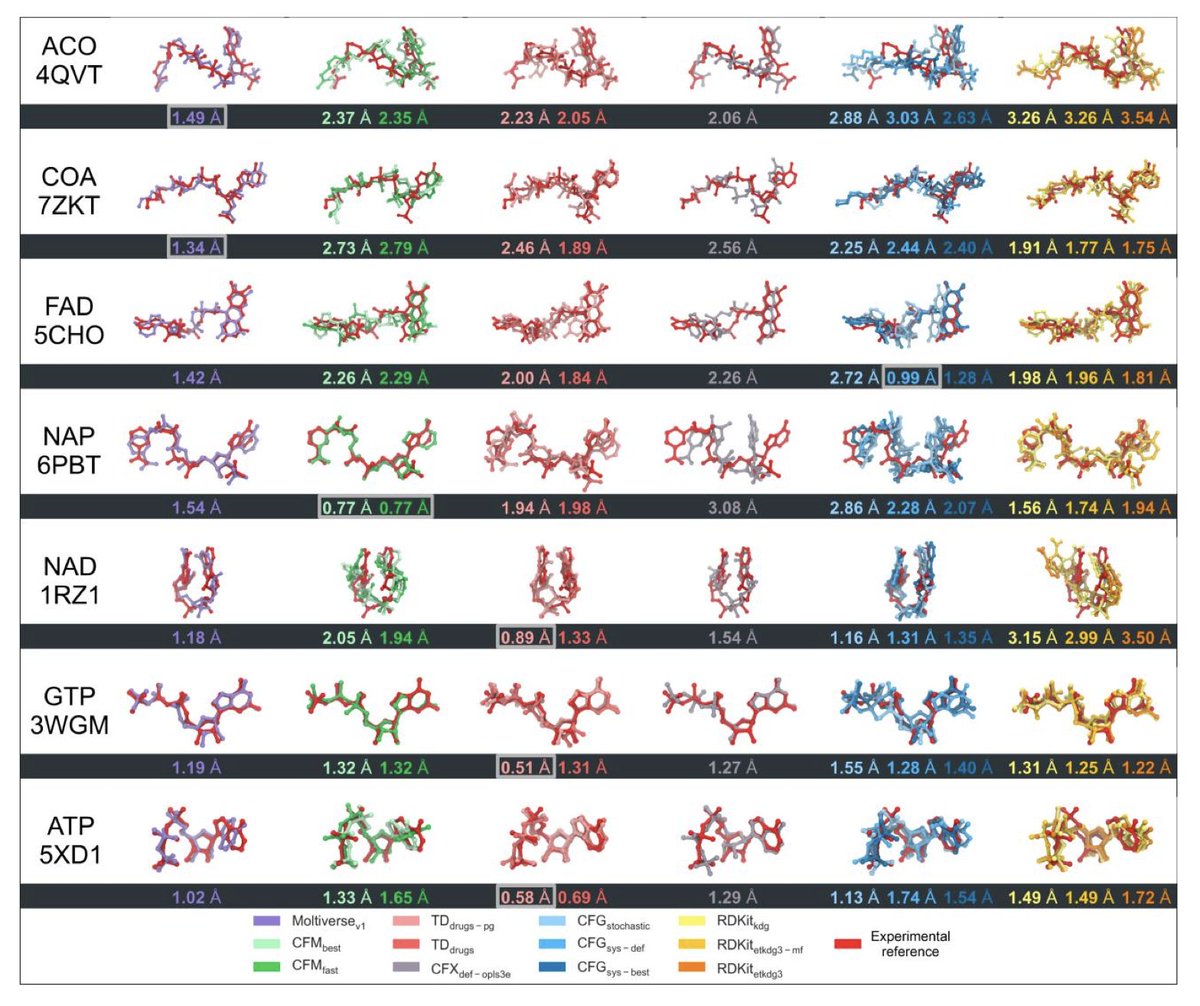
Moltiverse: Molecular Conformer Generation Using Enhanced Sampling Methods
1. Introducing Moltiverse: This novel protocol leverages enhanced sampling molecular dynamics, combining the extended adaptive biasing force (eABF) with metadynamics to efficiently explore molecular conformations. This approach enables Moltiverse to overcome energy barriers that commonly limit traditional molecular dynamics simulations.
2. Comprehensive benchmark: Moltiverse was evaluated against tools like RDKit, ConfGenX, and Torsional Diffusion, demonstrating competitive accuracy while maintaining high precision across diverse conformer generation benchmarks, including the newly introduced Cofactorv1 dataset.
3. Cofactorv1 dataset: Moltiverse is tested on Cofactorv1, a challenging dataset with seven small-molecule cofactors that feature extensive conformational diversity and high rotatable bond counts. This dataset presents a rigorous test for conformer generation tools, pushing the limits of current methodologies.
4. Structural diversity and accuracy: Moltiverse achieves low RMSD values and wide-ranging radii of gyration (RDGYR), indicating its ability to produce accurate, diverse conformations. Its hierarchical clustering step ensures that Moltiverse can deliver precisely the requested number of diverse conformers.
5. Computational efficiency: Moltiverse leverages a modern, streamlined protocol, including QM optimization with XTB software, enabling accurate conformations while keeping computational costs reasonable. Its implementation is optimized for parallel processing on standard CPUs, requiring no specialized hardware.
6. Broad applicability: Beyond conformer generation, Moltiverse holds promise for applications in drug discovery, where accurately modeled ligand conformations are critical for virtual screening and molecular docking.
@JansAlzate
💻Code: github.com/ucm-lbqc/moltiver…
📜Paper: doi.org/10.26434/chemrxiv-20…
#ConformerGeneration #ComputationalChemistry #DrugDiscovery #MolecularDynamics #EnhancedSampling
1
16
63
6,208
31 Oct 2024
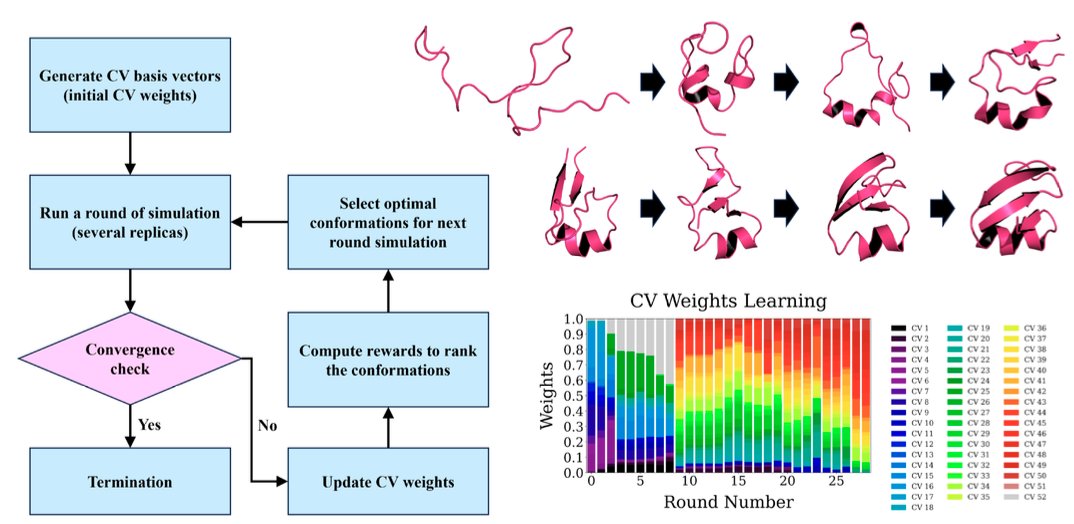
Adaptive CVgen: Leveraging Reinforcement Learning for Advanced Sampling in Protein Folding and Chemical Reactions @PNASNews
• Adaptive CVgen is a novel adaptive sampling framework that combines high-dimensional reaction coordinates with reinforcement learning (RL) to tackle the challenges in protein folding and complex chemical reactions.
• This method addresses two key challenges: identifying effective reaction coordinates and balancing exploration-exploitation to escape local energy minima, critical in protein and molecular dynamics.
• By dynamically generating and adjusting a comprehensive set of collective variables (CVs), Adaptive CVgen enables efficient, unbiased exploration of the entire conformational space without relying on predefined system knowledge.
• Reinforcement learning is utilized to optimize the CV weights in real-time, guiding sampling efficiently along the most probable evolutionary paths while adapting to dynamic system changes.
• The framework achieves high sampling efficiency, as demonstrated by successful folding of six proteins from disordered states to native conformations and accurate modeling of the C60 chemical synthesis process.
• This approach enables exploration of high-dimensional phase spaces with improved accuracy and efficiency, making it ideal for applications in drug discovery, protein dynamics, and molecular design.
📜Paper: pnas.org/doi/10.1073/pnas.24…
#ProteinFolding #ReinforcementLearning #Bioinformatics #EnhancedSampling #MolecularDynamics #ComputationalBiology
7
16
1,881

29 Aug 2024
🚨 New Paper 🚨
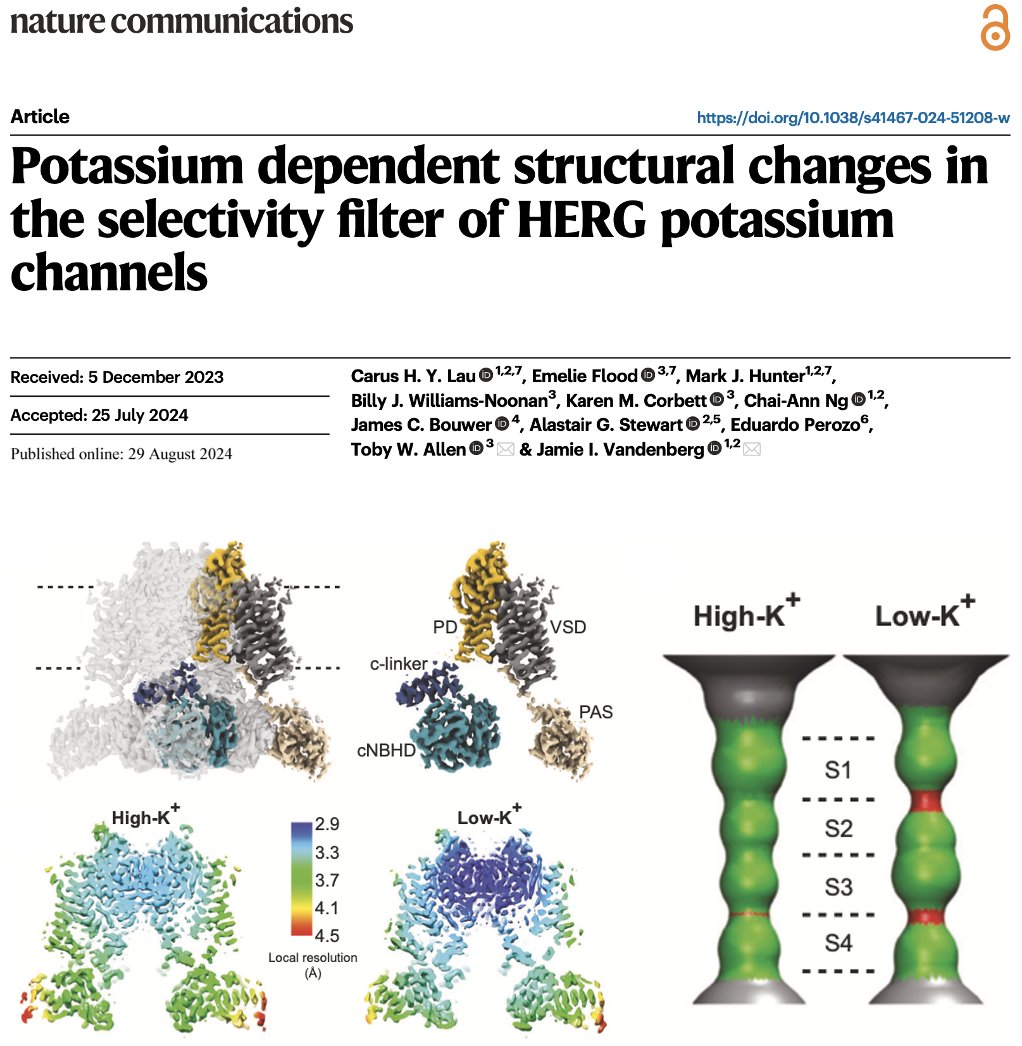
"Potassium dependent structural changes in the selectivity filter of #HERG potassium channels"
👇
nature.com/articles/s41467-0…
Well done @tobywilliamall1, @VandenbergJamie @EmelieJFlood @alastairstewart and everyone!
#EnhancedSampling #MolecularDynamics #HerG #CryoEM
29 Aug 2024

A significant effort to understand the rapid inactivation of hERG channels lead by @VandenbergJamie @CarusL Mark Hunter and our collaborators @tobywilliamall1 @EmelieJFlood @Dr_BillyWN Karen Corbett, @alastairstewart Eduardo Perozo. Exciting times ahead!
rdcu.be/dSl0r
2
1
2
921
16 Apr 2024
This letter (doi/10.1021/acs.jcim.4c00273) with Axel from @GroupParrinello is online in @JCIM_JCTC. Based on M06-2X, DeePKS, DeePMD, OPES and Voronoi CVs, we discovered a new proton transfer pathway for glycine tautomerism. #compchem #machinelearning #enhancedsampling
1
1
4
951