
Gene set signature scoring: great for quick #hypothesistesting and comparisons on any of the > 3,000 public resources (e.g. TCGA, DepMap, GTeX, etc..) that can be used instantly in r2platform.com
No bioinformatics / coding expertise needed
Cited in PubMed>3,200 times
1
73
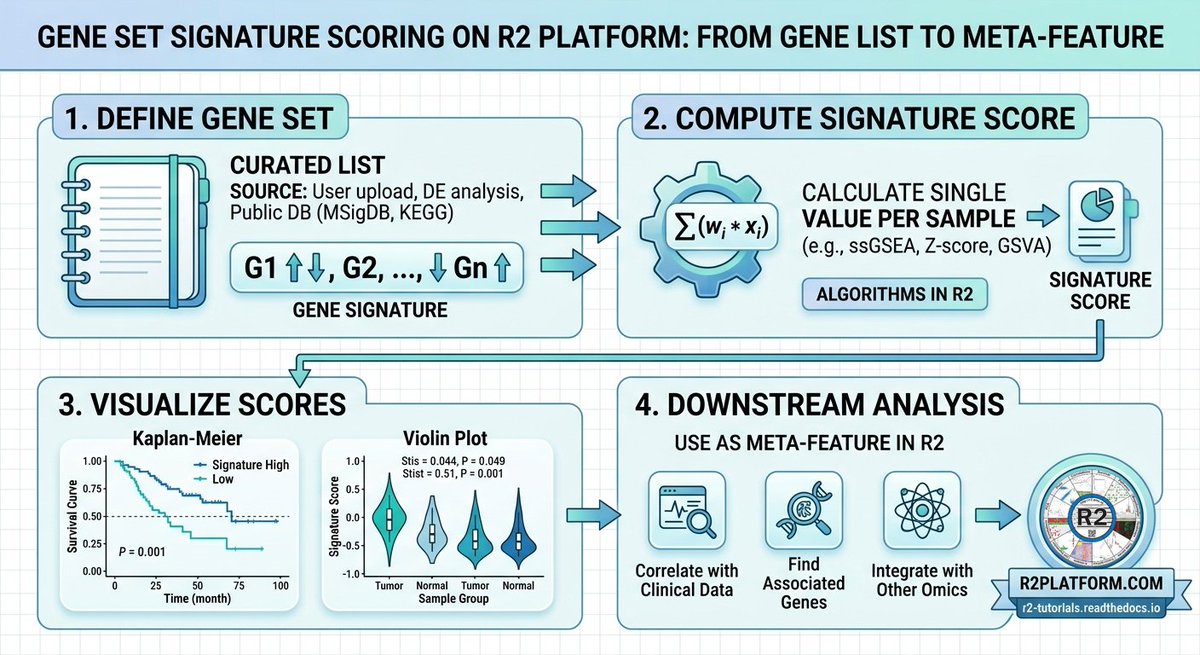
The open online R2 platform for biomedical researchers (r2platform.com) has a range of algorithms implemented to create and use gene set signature scores.
Great for quick #hypothesis testing on any of the > 3,000 public resources (e.g. TCGA, DepMap, GTeX, etc..)
..
1
1
4
110

Jun 12
A Molecular Glue That Cripples BRAF-Mutant Colorectal Cancer
Despite major advances in precision oncology, BRAF-mutant colorectal cancer (CRC) remains one of the most difficult gastrointestinal cancers to treat.
The problem is not the absence of BRAF inhibitors.
The problem is resistance.
Even approved combinations such as:
🧬 Encorafenib anti-EGFR
typically provide only short-lived benefit before MAPK signaling reactivates and tumors relapse.
A new Nature study introduces an entirely different strategy:
🎯 Destroy HuR (ELAVL1)
using a molecular glue degrader.
HuR emerges as a hidden vulnerability
HuR is an RNA-binding protein that regulates:
• mRNA stability
• pre-mRNA processing
• translation of oncogenic transcripts.
The authors designed a large CRBN-targeted molecular glue library and identified:
🧪 dHuR-1
followed by a more potent derivative:
🧪 dHuR-2.
These compounds recruit:
CRBN E3 ligase
→ HuR
→ ubiquitination
→ proteasomal degradation.
dHuR-2 achieved:
⚡ DC50 ≈ 3.8 nM
⚡ Dmax ≈ 96%
with rapid and selective HuR degradation.
Structural biology reveals a new molecular glue mechanism
Cryo-EM analysis solved the:
🧬 CRBN–dHuR-2–HuR ternary complex
at 3.3 Å resolution.
The degrader creates a novel interaction surface that recruits:
🎯 HuR G58 β-hairpin degron
onto CRBN.
Mutation of:
G58
completely abolished degradation.
This establishes HuR as a bona fide molecular glue substrate.
BRAF-mutant CRC is uniquely sensitive
Analysis of DepMap datasets revealed a striking observation:
BRAF mutation status was the strongest predictor of HuR dependency.
Across CRC models:
🟢 All BRAF-mutant lines responded strongly
⚪ BRAF-wildtype lines were largely resistant.
Genetic deletion of HuR reproduced the phenotype, confirming on-target dependency.
The most surprising mechanism: alternative splicing
The study uncovered a completely unexpected pathway.
HuR binds a:
🧬 U-rich element
within BRAF intron 17.
Under normal conditions:
HuR
→ preserves inclusion of exon 18
→ generates functional BRAF transcript.
After HuR degradation:
❌ Exon 18 is skipped
→ abnormal BRAF isoform (BRAF-X2)
→ impaired translation
→ dramatic reduction of BRAF protein.
The result is sustained collapse of MAPK signaling.
Dual suppression of BRAF and EGFR
Unlike conventional BRAF inhibitors that trigger:
⚠ EGFR feedback activation
⚠ ERK rebound signaling
⚠ resistance within hours
HuR degradation simultaneously reduced:
⬇ BRAF
⬇ EGFR
⬇ p-MEK
⬇ p-ERK.
Importantly, MAPK suppression remained durable for days rather than hours.
Activity persists in BRAF inhibitor–resistant tumors
One of the strongest findings involved resistant models.
Investigators generated:
🧬 Dabrafenib-resistant CRC
and melanoma lines.
Despite resistance to:
• Dabrafenib
• Encorafenib
HuR degraders remained highly active.
The resistant tumors still depended on:
HuR
→ BRAF
→ MAPK signaling.
Thus the degrader bypasses one of the most important resistance mechanisms in BRAF-driven cancers.
Synergy with existing targeted therapies
The authors next evaluated combinations.
dHuR BRAFi
dHuR EGFR inhibitor
dHuR MEK inhibitor
all produced strong synergistic effects.
A kinome-wide CRISPR screen independently identified:
🎯 EGFR
🎯 MAPK1 (ERK2)
among the top synthetic-lethal partners of HuR degradation.
This reinforces the central role of MAPK pathway suppression.
Anti-angiogenic activity adds another layer
HuR degradation also reduced:
🧬 VEGFA
through disruption of VEGFA RNA processing.
In xenografts:
⬇ CD31-positive vessels
⬇ microvessel density
were observed after treatment.
Thus HuR degradation attacks tumors through:
1️⃣ BRAF suppression
2️⃣ EGFR suppression
3️⃣ VEGFA suppression
simultaneously.
Why this matters
This study introduces a new therapeutic paradigm.
Instead of inhibiting:
• BRAF kinase activity
or
• EGFR signaling
the strategy eliminates an upstream RNA-binding regulator that controls both.
The proposed pathway becomes:
HuR degradation
→ BRAF exon 18 skipping
→ loss of BRAF protein
→ suppression of MAPK signaling
→ reduced tumor growth
→ overcoming BRAFi resistance.
The work also expands the molecular glue field beyond transcription factors and kinases into:
🧬 RNA-binding proteins
a class long considered difficult to drug.
With the clinical candidate DEG6498 already entering clinical development, this may represent one of the first examples of targeting oncogenic RNA processing through molecular glue–induced protein degradation.
Reference
Lu X, Wang X, Yang Z, et al.
Molecular glue degraders of HuR suppress BRAF-mutant colorectal cancer.
Nature (2026)
DOI: 10.1038/s41586-026-10613-5
#ColorectalCancer #BRAF #HuR #ELAVL1 #MolecularGlue #TargetedProteinDegradation #CRBN #PrecisionOncology #CancerTherapeutics #NatureJournal

1
7
347
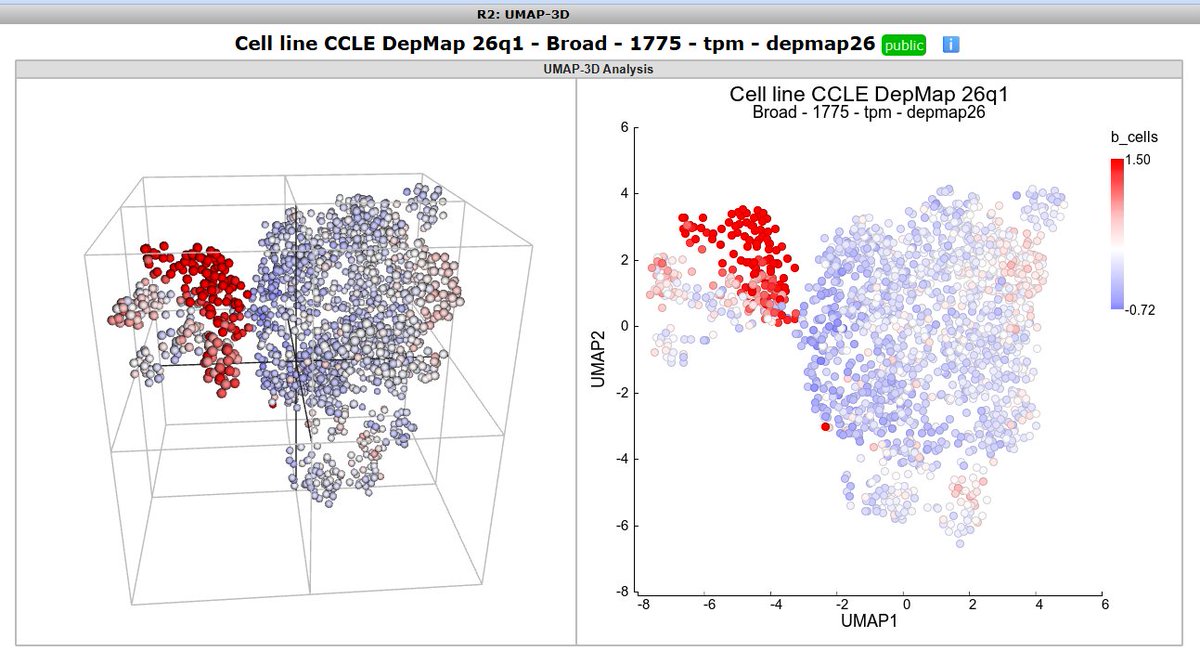
3d-UMAP one of the cool functions in the open online R2 platform ( r2platform.com ) for playful #hypothesistesting in e.g. #DepMap
#dataviz #umap
1
46

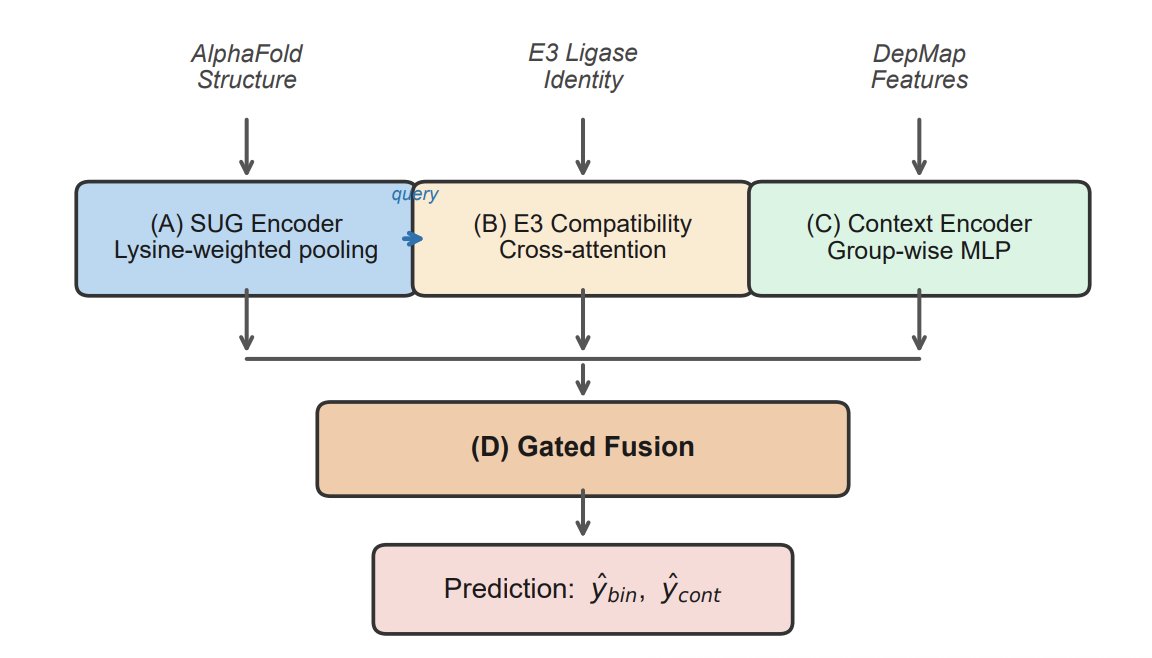
Structure-Aware Prediction of PROTAC-Mediated Protein Degradability via Graph Neural Networks
1. DegradoMap targets a key early-stage bottleneck in PROTAC discovery: predicting whether a protein is degradable before any PROTAC is designed. It uses only an AlphaFold structure (via UniProt ID) plus the E3 ligase identity, avoiding the “need the full PROTAC structure” requirement of many prior predictors.
2. The core idea is to encode biophysical priors of ubiquitin transfer directly into a structure GNN. The model builds a residue graph (8Å radius) and applies an SE(3)-invariant message passing network that uses distances (not orientations) to predict a scalar degradability probability.
3. A central architectural innovation is lysine-weighted graph pooling with per-protein softmax normalization plus protein-size normalization. This focuses representations on ubiquitination-relevant residues while preventing subtle leakage and “protein size shortcuts”; these fixes substantially improved target-unseen generalization in ablations.
4. To model protein–E3 compatibility, DegradoMap adds a bidirectional cross-attention module between the protein representation and a learned E3 embedding (10 ligases in the benchmark). This is designed to capture E3-specific geometric/steric constraints that influence ternary complex formation.
5. The model also integrates cellular context using 59 Cancer Dependency Map (DepMap) features (expression, dependency, CNV, proteomics, mutations, etc.), fused with structure and E3 signals via a learned gating mechanism; training is multi-task (binary degradability continuous efficiency).
6. On PROTAC-8K (3,101 samples; 155 targets; 10 E3s), performance depends strongly on evaluation protocol. Target-unseen testing (proteins never seen in training) is emphasized as the realistic target-selection setting: mean AUROC 0.646 ± 0.124 (3 seeds), but 0.603 ± 0.097 over 6 seeds, highlighting large seed variance and the need for ensembling.
7. Cross-E3 transfer was strong in an E3-unseen setup (train on CRBN, test on VHL): AUROC 0.811, suggesting the model can learn structural signals that transfer across ligases under that axis of generalization.
8. Beyond classification, DegradoMap can recommend which E3 ligase to try: Hit@3 = 74% (Hit@1 = 46%, MRR = 0.641) on a subset with known E3 preferences, aiming to reduce the cost of parallel E3 exploration.
9. Two broader ML takeaways are reported: (i) E(3)-equivariant architectures underperformed the simpler invariant design for this scalar prediction task, and (ii) ESM-2 residue embeddings only helped with careful regularization—naive integration degraded performance.
10. Practical reliability is addressed via calibration: on the target-unseen split, confidence scores were well-calibrated (ECE = 0.029), enabling threshold-based screening. Key limitations include E3 imbalance in data (CRBN/VHL dominate), poor target-unseen performance on VHL-targeted proteins (AUROC 0.396), and the absence of external benchmarks.
💻Code: github.com/bryanc5864/Degrad…
📜Paper: arxiv.org/abs/2606.04021
#PROTAC #TargetedProteinDegradation #GNN #GeometricDeepLearning #AlphaFold #ComputationalBiology #DrugDiscovery #MachineLearning
7
17
1,517

Jun 4
Whole genome perturbations such as whole genome perturb seq and DepMap are rare examples of published negative results (i.e we have the data for the cells where the KO does nothing).
There is plenty of evidence from those data that our best model suck.
Because of positive bias.
Jun 3

Most experiments fail, and negative results rarely get published. This means LLMs are unaware of the outcomes of most experiments.
2
59

May 31
surely 99% of interesting released datasets were generated in academia
sure perhaps via a consortium but that’s still academia. Eg Broad depmap
If ur point is just make more consortia sure maybe but I think those exist bc it’s easier for 50 professors to get funded than 1. Coordination overhead is insanely high tho.
I think it’d be easier to just require data release and let smaller/more groups do it.
3
4
533

ほぼPCだけでできるインシリコ創薬論文紹介10
DepMap公開データでがんの弱点を探す依存性マップ研究
academic.oup.com/gigascience…
pubmed.ncbi.nlm.nih.gov/3799…
pubmed.ncbi.nlm.nih.gov/4113…

2
14
1,584

A copy number of 4 in DepMap does not mean what you think it means.
Half the analyses I see treat OmicsCNGene.csv like it contains absolute copy number. It does not.
1
1
10
4,438

Apr 28
@grok verify each claim individually and as a whole: (also add any value if and only if you are fully certain in your value add approach)
This is the human equivalent of the landmark yeast genetic interaction maps from the same group (Costanzo et al. 2010, 2016).
~89,000 interactions across ~4M gene pairs in HAP1 cells, revealing that the hierarchical modular topology of genetic networks — complexes → pathways → processes → compartments — is conserved from yeast to human despite ~1B years of divergence, even though only ~7% of individual interactions are shared between ortholog pairs.
- The key insight for translational biology: this network captures functional relationships among nonessential genes with subtle fitness effects that DepMap co-essentiality completely misses (Jaccard ~0.1 at stringent thresholds).
- The ~1,800 suppression interactions are a goldmine for identifying disease gene modifiers — the TAFAZZIN-ABHD18 Barth syndrome example already validated in vivo underscores this.
Apr 27

Now online! Global genetic interaction network of a human cell maps conserved principles and informs functional interpretation of gene co-essentiality profiles dlvr.it/TSFljT
1
2
40

#AACR26 @AACR · Tools and platform layer: spatial biology, DepMap, AI pathology
@BiotechTV on 10x Genomics @MaverickNY on Bill Sellers' DepMap talk @IamLinghua's MD Anderson collaboration thread — the platform-layer signals form a coherent picture of the infrastructure underlying the clinical narratives.
10x Genomics highlighted Atera, a new in situ spatial biology platform, at #AACR26; Serge Saxonov (10x co-founder/CEO) discussed the platform's goals around higher-throughput, higher-resolution spatial multi-omics (per @BiotechTV).
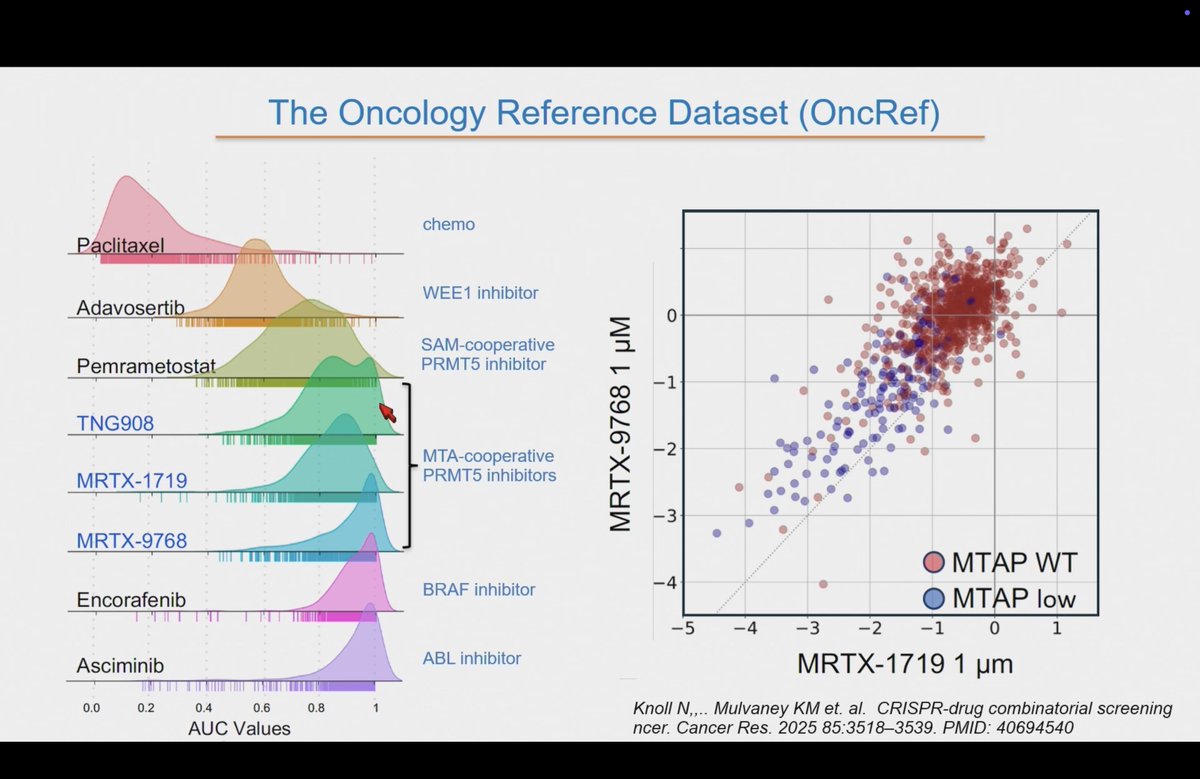
Bill Sellers (@broadinstitute) discussed future directions with DepMap, including differentiated pharmacology between PRMT5 inhibitors — SAM-cooperative vs MTA-cooperative inhibitors showing distinct selectivity profiles, illustrating how functional genomics can distinguish between mechanistically similar molecules (per @MaverickNY; Cottrell et al., J Med Chem 2025).
Linghua Wang @IamLinghua (@UTMDAnderson) reported multiple collaborative (@humam_kadara @PavlosMsaouel @tae_hwang @JeremyLDavisMD @DrMingyaoLi @MansfieldPaul @JafferAjaniMD) posters at #AACR26 on AI-augmented pathology and spatial omics convergence, with @UTMDAnderson highlighting how AI pathology and spatial omics can jointly decode tissue architecture.
The unifying thread: these platforms are approaching a resolution where the translational bottleneck shifts from data generation to hypothesis selection — a structural shift the field has been anticipating.
x.com/BiotechTV/status/20456…
x.com/MaverickNY/status/2045…
x.com/IamLinghua/status/2045…

Also excited to highlight 12 posters from our collaborative teams at #AACR26 @humam_kadara @PavlosMsaouel @tae_hwang @JeremyLDavisMD @DrMingyaoLi @MansfieldPaul @JafferAjaniMD Drs. Gao & Maru. Grateful for these outstanding collaborations—don’t miss them!

1
5
1,400

Apr 18
Excellent talk from Bill Sellers on future directions with DepMap. Interesting differences between different PRMT5 inhibitors (SAM & MTA) and how they compare with selective inhibitors #aacr26
1
7
50
5,573
Updated resource: The versatile r2platform.com now also includes the latest 2026q1 version of #DepMap. Explore this superb resource also in the R2 ecosystem. e.g. use data driven embeddings to explore the composition of the 1775 cancer cell lines.
3000 public resources
1
3
115

Attending @AACR? Stop by these Cancer Dependency Map activities:
Saturday, April 18; 12:30 - 2:00 pm; Ballroom 6 DE - Upper Level - Convention Center: Join the DepMap team in celebrating their 2026 AACR Team Science Award at the Award Lecture. aacr.org/about-the-aacr/news… (2/4)
1
2
853

Congratulations to @boehmjesse, our Chief Science Officer, and the @broadinstitute DepMap team for being among the honorees of the 2026 @AACR Scientific Achievement Awards.
This team is recognized for their work in systematically mapping genetic dependencies across cancer cells, uncovering lineage- and genotype- specific cancer vulnerabilities.
Join us in congratulating the DepMap team for this well deserved recognition
aacr.org/about-the-aacr/news…
2
7
417
Test the genes as a result of your Claude prompt in >550,000 bulk samples with ease in R2: the open online data science platform 4 biomedical research r2platform.com
R2 provides instant access to nearly 3,000 public resources including #TCGA, #GTeX, #DepMap, target, etc
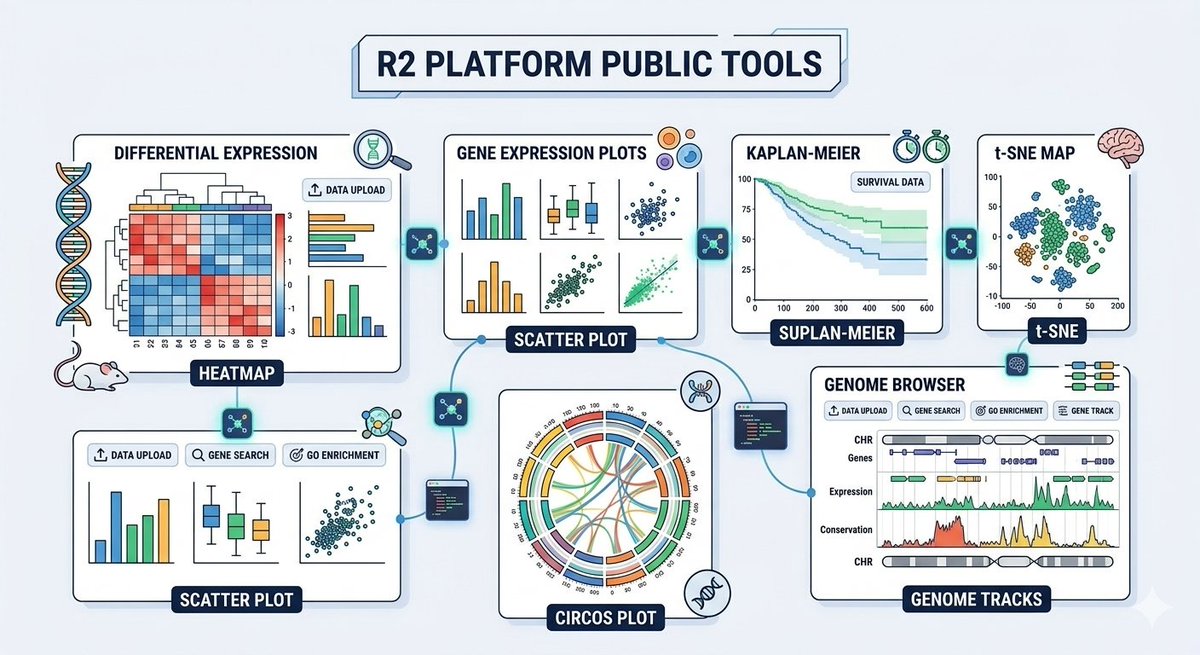
4
288
Explore 3.000 public resources (500.000 bulk & 5 million single dells) instantly in the open online data #science platform r2platform.com . Designed for biomedical researchers.
e.g. GTeX
TCGA, DepMap, Atlasses and many more
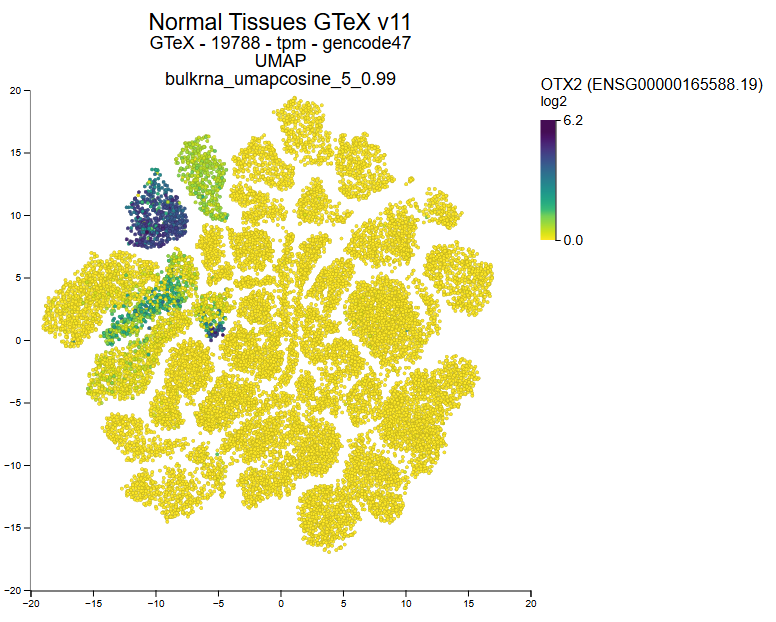
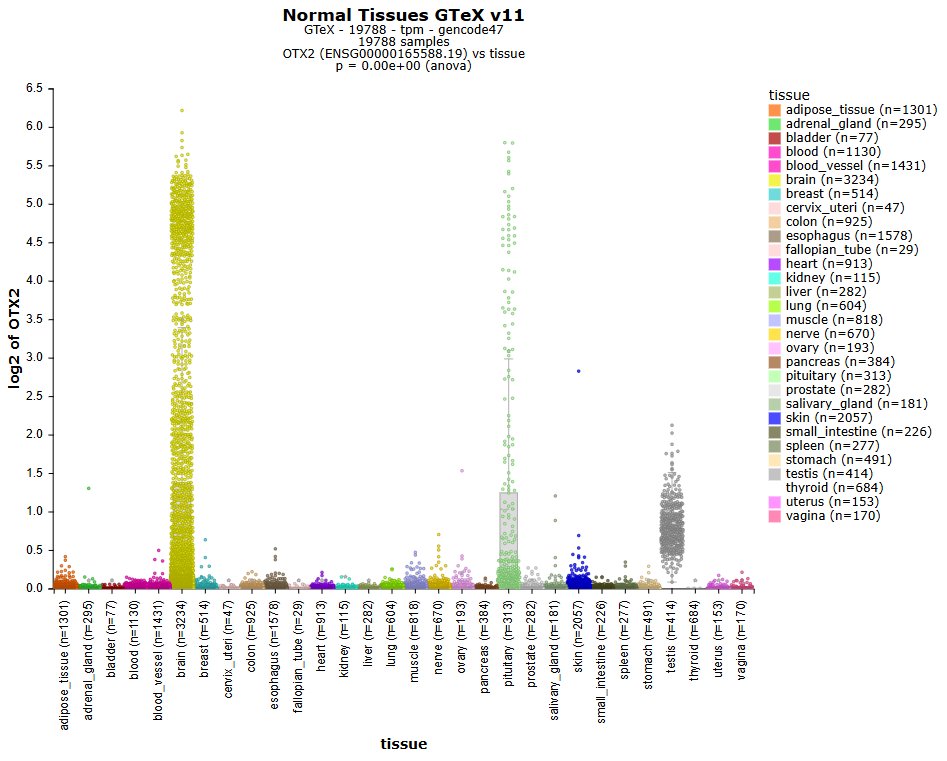
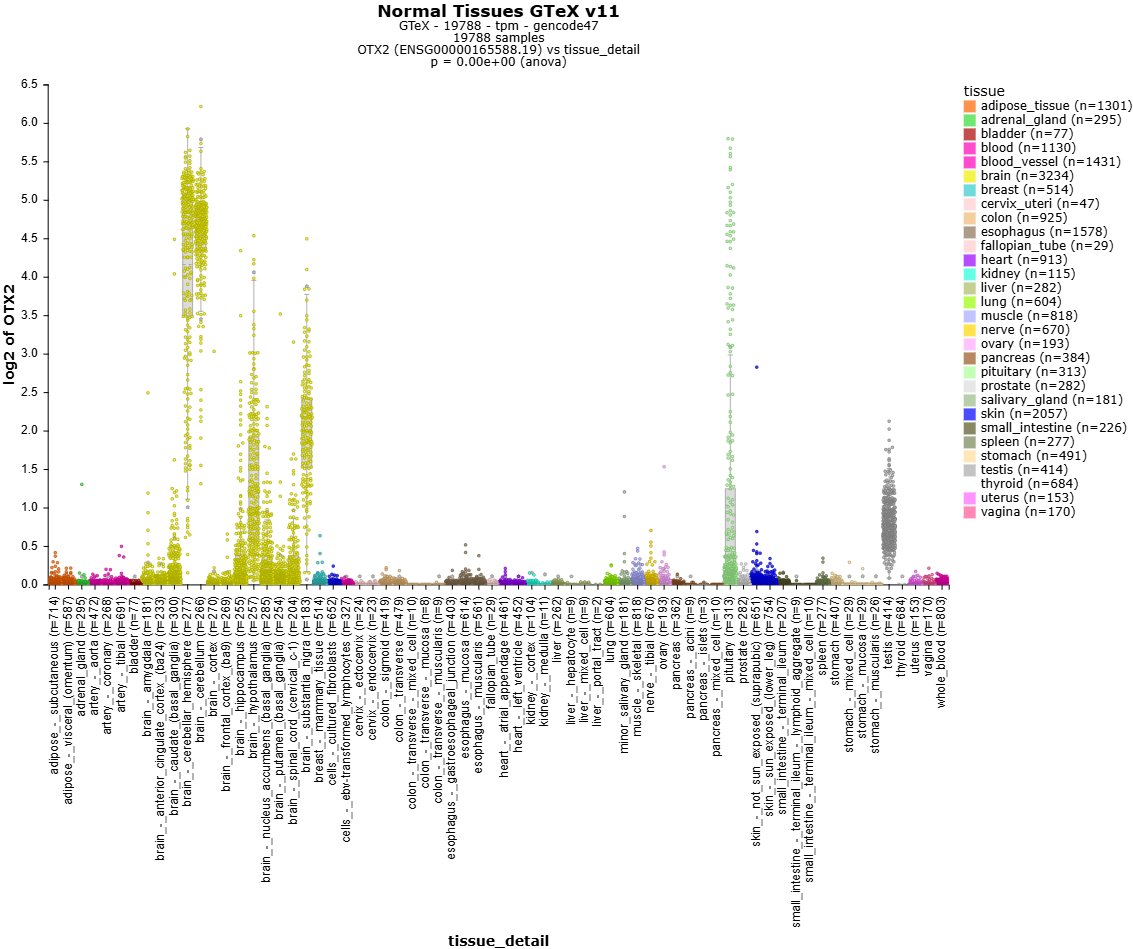
2
3
121
Test the genes as a result of your openAI ChatGPT prompt in >500,000 bulk samples with ease in R2: the open online data platform for biomedical #research r2platform.com
R2 provides instant access to nearly 3,000 public resources including TCGA, GTeX, DepMap, target, etc

1
3
121

Mar 19
Recursion is headed to #AACR26 - April 17-22 in San Diego - to share our breakthroughs in leveraging data and AI to drive new cancer discoveries and treatments.
🔹 On April 18, 12:30pm, Teeru Bihani, VP of Translational Strategy at Recursion, will present as part of an educational session on "AI in Biomarker Discovery and Translational Drug Development" along with Jorge Reis-Filho, Chief of AI for Science Innovation at @AstraZeneca. She’ll share details of Recursion’s AI-enabled cancer programs, including REC-1245, in which we used Recursion’s AI platform to discover the relationship between RBM39 and DNA damage response, and to design a potentially first-in-class degrader which is being developed for the treatment of biomarker-enriched solid tumor indications and lymphoma. The session will be moderated by Jakob N. Kather of Dresden Graduate School for Interdisciplinary Life Sciences.
🔹 On April 20, 9am-12pm, we’ll present a poster on CellNeighbor, a transcriptional atlas of patient tumors and cell line models to inform preclinical model selection. CellNeighor addresses the translational gap between cell line models selected for compound activity assays based on molecular profiles and the in vitro cell culturing that can influence genetic changes in these models. We built CellNeighbor by integrating transcriptomic profiles from DepMap with patient tumor data from The Cancer Genome Atlas and deidentified patient tumor data from @TempusAI, creating a unified transcriptomic map of cell lines and tumors.

1
3
21
4,362

Scaling alone isn’t enough.
We explicitly inject biological knowledge via cross-attention:
-- protein language models
-- gene embeddings from text
-- interaction networks (STRING)
-- dependency maps (DepMap)
-- morphology profiles
This lets the model move beyond pattern matching → mechanistic reasoning.
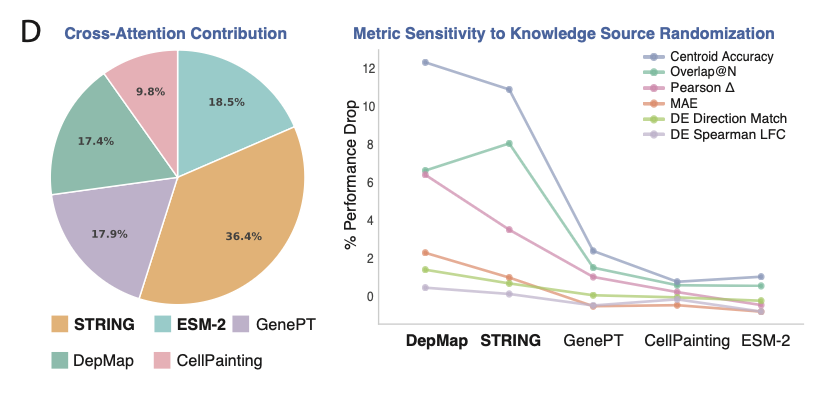
1
1
17
3,627