
An electron-density point-cloud framework for robust protein-ligand interaction prediction
1. E-CloudBind reframes protein–ligand affinity prediction around electron-density point clouds rather than relying on sub-ångström atomic coordinates, aiming to stay accurate when structures are low-resolution or predicted (e.g., AlphaFold) and therefore noisy.
2. Key idea: replace hard distance cutoffs for “contacts” (e.g., within 5 Å) with density-aware Gaussian “electron clouds”, where interactions are defined by overlap/isosurface intersection, yielding a more resolution-agnostic interaction graph.
3. The framework explicitly splits chemistry into two complementary channels: non-covalent interactions from 3D electron-cloud point clouds, and covalent structure from intrinsic molecular graphs (bond topology), then fuses them for affinity regression.
4. Ligand electron density is obtained via semi-empirical quantum chemistry (GFN2-xTB), while protein pockets use a van der Waals radius-guided multivariate Gaussian sampling strategy as a physically motivated proxy that is far cheaper than full QM density.
5. Architecture highlights: K-means clusters point clouds into atom-aligned local regions; a point-cloud encoder (3D-GCN-style deformable kernels) learns local non-covalent patterns (e.g., H-bonds, π-stacking, van der Waals complementarity); a heterogeneous GNN encodes covalent graphs; a multi-bond fusion module integrates both.
6. On PDBbind 10-fold CV, E-CloudBind reports MAE 1.059 and Pearson 0.667, outperforming representative sequence-based (PSICHIC), graph-based (SIGN), and structure-based (DMFF) baselines, and also comparing favorably to recent structure-centric methods (EHIGN, Boltz-2, FlowDock) under the same protocol.
7. Robustness to experimental resolution: when regressing absolute error vs. crystallographic resolution, E-CloudBind shows a much flatter slope (0.017) than baselines (0.053–0.065), with a non-significant trend (p = 0.703), indicating reduced sensitivity to declining structural quality.
8. Robustness to structure source shifts: swapping experimental proteins with AlphaFold2 models causes only a small performance change for E-CloudBind (MAE 0.042; Pearson −0.004), while coordinate-dependent baselines degrade more (e.g., DMFF MAE 0.187; Pearson −0.093).
9. Out-of-distribution testing built from DAVIS via combinatorial partitioning by protein/ligand complexity shows tighter error dispersion for E-CloudBind (lowest median deviation), with stable performance across increasing protein Relative Contact Order and ligand Bertz complexity.
10. Practical and interpretability results: attention maps highlight polar ligand atoms and key pocket regions consistent with known interaction motifs; large-scale screening on 80,383 ZINC molecules against PBP1A, SARS-CoV-2 Mpro, and BCL-2 uses docking for follow-up, plus BCL-2 candidate assessment with synthesizability metrics and explicit-solvent MD (400 ns) suggesting stable binding for selected hits.
💻Code: github.com/Liuyujian0408/DPI ; doi.org/10.5281/zenodo.19851…
📜Paper: doi.org/10.1038/s41467-026-7…
#ComputationalBiology #DrugDiscovery #ProteinLigand #BindingAffinity #GeometricDeepLearning #GNN #PointCloud #ElectronDensity #VirtualScreening #AlphaFold

2
23
1,379
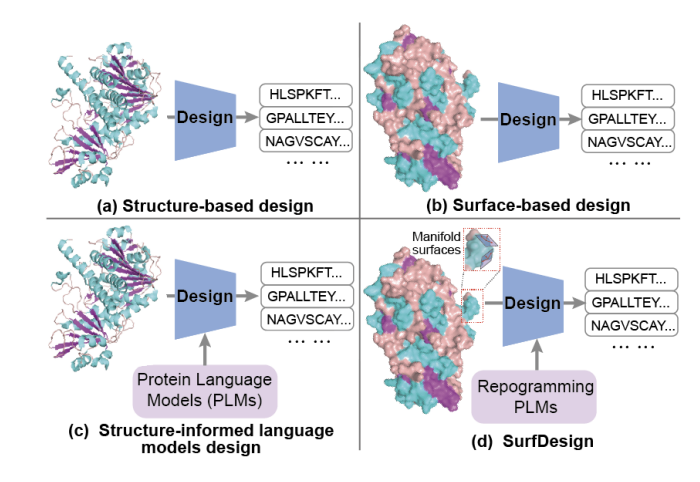
SurfDesign: Effective Protein Design on Molecular Surfaces
1. SurfDesign reframes protein design around molecular surfaces (shape physicochemical complementarity), aiming to better control functional regions like binding interfaces and enzyme pockets compared with backbone-only conditioning.
2. Core technical idea: treat molecular surfaces as continuous geometric manifolds rather than unordered point clouds/meshes, so the model can leverage local tangent structure, curvature, and directional consistency that are important for interaction-centric design.
3. The method builds an oriented surface point cloud Q where each point includes coordinates, a unit normal vector, and physicochemical attributes (e.g., hydrophobicity, charge, H-bond features). Surfaces are generated via PyMOL/MSMS and denoised with Gaussian smoothing; no residue identities, MSAs, or functional labels are used in surface generation.
4. SurfDesign introduces a Surface-conditioned Equivariant Message Passing (SEMP) encoder: SE(3)-equivariant updates use invariant radial distances, curvature descriptors (from local covariance eigenvalues), and directional angular features derived from surface normals (two intersection angles one dihedral angle).
5. Directionality is encoded with spherical Fourier–Bessel bases over distances and angles; messages are attention-reweighted and used to update both node features and coordinates, with optional per-layer recomputation of normals/curvatures to stay consistent as coordinates evolve.
6. To address limited surface-structure paired data and improve sequence priors, SurfDesign integrates pretrained protein language models (PLMs) via parameter-efficient fine-tuning (hybrid PEFT: structural adapter LoRA), trained with conditional masked language modeling rather than autoregressive decoding.
7. Binder design benchmark (6 targets) uses AF2 pAE_interaction as a functional proxy. SurfDesign achieves the best average pAE_interaction (15.85) and the highest overall success rate (30.14%), outperforming SurfPro (surface-conditioned baseline) and backbone-only baselines like ProteinMPNN, PiFold, and LM-DESIGN.
8. Enzyme design benchmark (5 enzyme–substrate systems; leakage-controlled by excluding overlaps with CATH pretraining) uses ESP score as a proxy for enzyme–substrate compatibility. SurfDesign attains the best average success rate (47.30%) and the best average ESP under greedy decoding (0.9058), with gains persisting in a zero-shot substrate setting.
9. Inverse folding is positioned as a diagnostic for structural compatibility (not “recovering the native sequence”). SurfDesign reports strong results on CATH splits, including perplexity 2.41 and AAR 74.13% on CATH 4.2, plus improved surface recovery metrics (IoU/CD/NC) versus PLM-based baselines; scaling larger PLMs further improves recovery.
💻Code: github.com/smiles724/SurfDes…
📜Paper: arxiv.org/abs/2606.07567
#ProteinDesign #ComputationalBiology #GeometricDeepLearning #ProteinEngineering #EnzymeDesign #ProteinBinding #EquivariantNetworks #ProteinLanguageModels #KDD2026
15
74
3,918
Structure-Aware Prediction of PROTAC-Mediated Protein Degradability via Graph Neural Networks
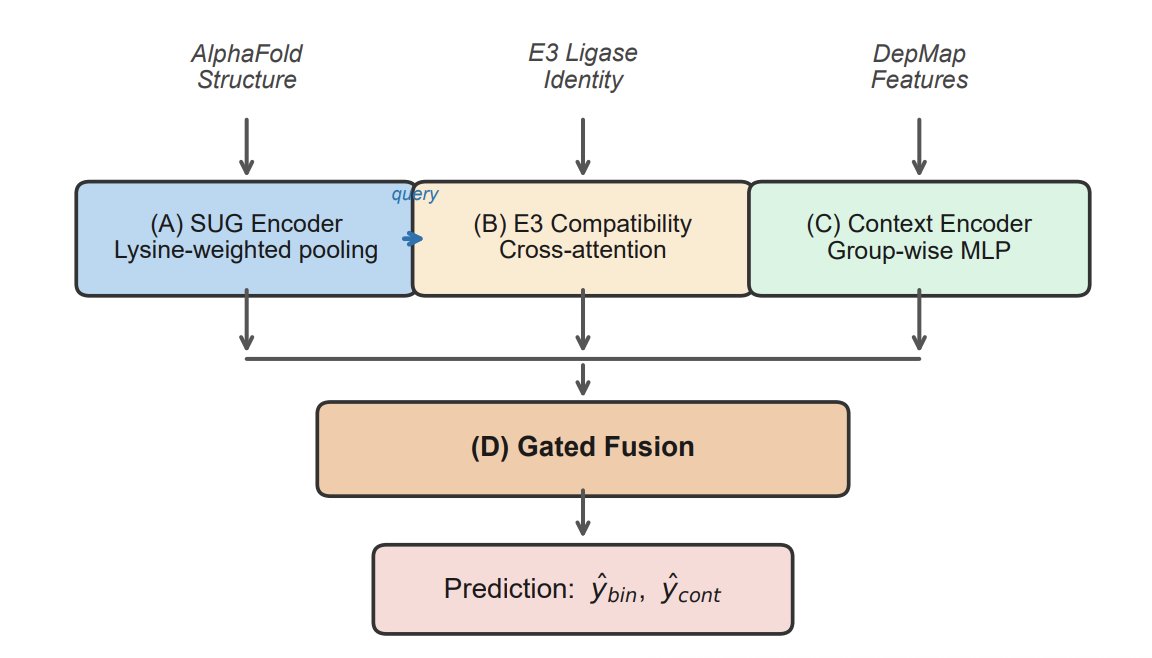
1. DegradoMap targets a key early-stage bottleneck in PROTAC discovery: predicting whether a protein is degradable before any PROTAC is designed. It uses only an AlphaFold structure (via UniProt ID) plus the E3 ligase identity, avoiding the “need the full PROTAC structure” requirement of many prior predictors.
2. The core idea is to encode biophysical priors of ubiquitin transfer directly into a structure GNN. The model builds a residue graph (8Å radius) and applies an SE(3)-invariant message passing network that uses distances (not orientations) to predict a scalar degradability probability.
3. A central architectural innovation is lysine-weighted graph pooling with per-protein softmax normalization plus protein-size normalization. This focuses representations on ubiquitination-relevant residues while preventing subtle leakage and “protein size shortcuts”; these fixes substantially improved target-unseen generalization in ablations.
4. To model protein–E3 compatibility, DegradoMap adds a bidirectional cross-attention module between the protein representation and a learned E3 embedding (10 ligases in the benchmark). This is designed to capture E3-specific geometric/steric constraints that influence ternary complex formation.
5. The model also integrates cellular context using 59 Cancer Dependency Map (DepMap) features (expression, dependency, CNV, proteomics, mutations, etc.), fused with structure and E3 signals via a learned gating mechanism; training is multi-task (binary degradability continuous efficiency).
6. On PROTAC-8K (3,101 samples; 155 targets; 10 E3s), performance depends strongly on evaluation protocol. Target-unseen testing (proteins never seen in training) is emphasized as the realistic target-selection setting: mean AUROC 0.646 ± 0.124 (3 seeds), but 0.603 ± 0.097 over 6 seeds, highlighting large seed variance and the need for ensembling.
7. Cross-E3 transfer was strong in an E3-unseen setup (train on CRBN, test on VHL): AUROC 0.811, suggesting the model can learn structural signals that transfer across ligases under that axis of generalization.
8. Beyond classification, DegradoMap can recommend which E3 ligase to try: Hit@3 = 74% (Hit@1 = 46%, MRR = 0.641) on a subset with known E3 preferences, aiming to reduce the cost of parallel E3 exploration.
9. Two broader ML takeaways are reported: (i) E(3)-equivariant architectures underperformed the simpler invariant design for this scalar prediction task, and (ii) ESM-2 residue embeddings only helped with careful regularization—naive integration degraded performance.
10. Practical reliability is addressed via calibration: on the target-unseen split, confidence scores were well-calibrated (ECE = 0.029), enabling threshold-based screening. Key limitations include E3 imbalance in data (CRBN/VHL dominate), poor target-unseen performance on VHL-targeted proteins (AUROC 0.396), and the absence of external benchmarks.
💻Code: github.com/bryanc5864/Degrad…
📜Paper: arxiv.org/abs/2606.04021
#PROTAC #TargetedProteinDegradation #GNN #GeometricDeepLearning #AlphaFold #ComputationalBiology #DrugDiscovery #MachineLearning
7
16
1,509
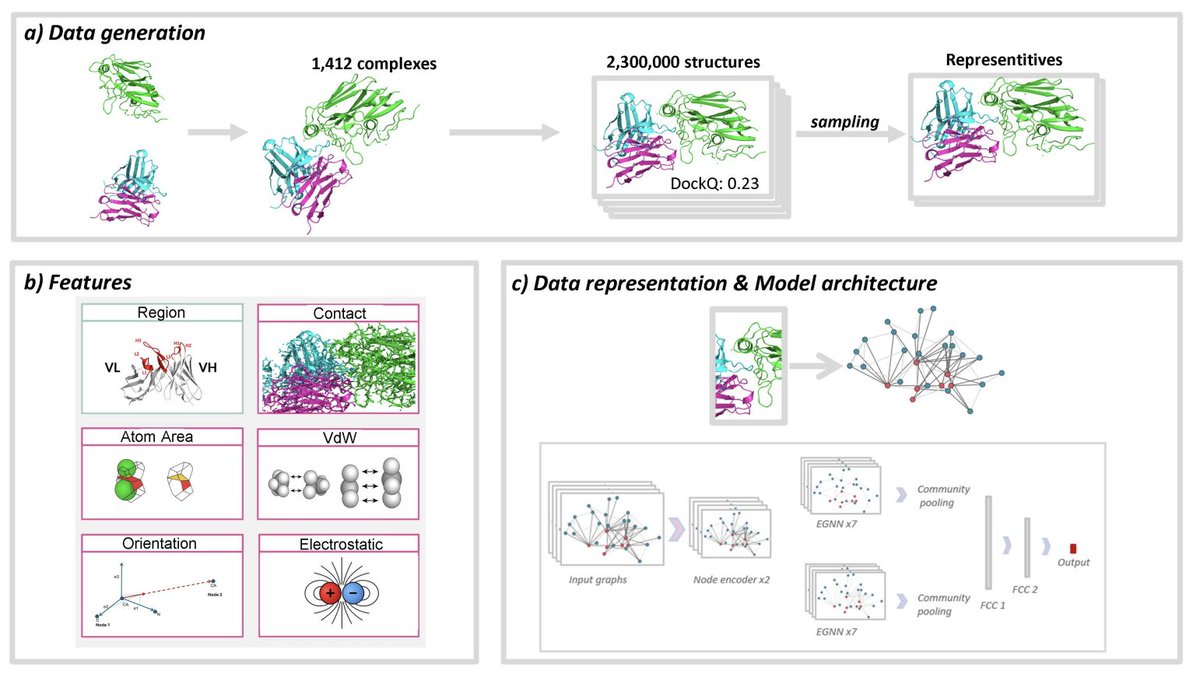
DeepRank-Ab: A scoring function for antibody-antigen complexes based on geometric deep learning
1. DeepRank-Ab targets a key failure mode in antibody–antigen modeling: good poses are often sampled but not ranked correctly. The paper shows an example where AlphaFold3 generates a DockQ 0.6 model yet ranks it 498th, illustrating that scoring (not only sampling) is a central bottleneck.
2. The authors introduce a rigorously curated benchmark tailored to antibody–antigen interfaces: ~2.3 million decoys from 1,442 complexes (from SAbDab), generated under four HADDOCK3 protocols (bound/unbound docking and refinement). Complexes are clustered with Foldseek-Multimer and split at the cluster level to reduce data leakage.
3. DeepRank-Ab is trained as a regression model to predict continuous DockQ (pDockQ), rather than a binary “acceptable/not acceptable” label. This preserves the full spectrum of model quality and directly supports ranking.
4. A major design finding: atom-level interface graphs outperform residue-level graphs for antibody–antigen scoring, consistent with the importance of fine-grained CDR loop geometry. The best-performing formulations combine atom-level nodes with interface-focused edges.
5. The interface representation is strengthened using Voronoi-based surface decomposition: edges can encode either simple interatomic distances or Voronoi contact areas. Across cross-validation, Voronoi contact area provides a consistent edge over distance-only encoding for predicting and classifying near-native poses.
6. Antibody-specific and physics-inspired features matter. Adding IMGT-derived region labels (e.g., CDR annotations) and geometric/energetic descriptors improves performance and stability; ablations show large error increases when removing region labels, atom type, covalent interactions, orientation, or Voronoi contact area. In contrast, raw ESM-2 embeddings are partly redundant, and antibody-specific fine-tuning (AbTune) yields only marginal gains relative to added complexity.
7. Training data sampling is treated as a first-class modeling choice. Besides “balanced” sampling across DockQ bins, they test targeted upsampling of low-quality decoys (DockQ < 0.23) to increase structural diversity in failure modes; this slightly improves overall learning and robustness.
8. Architecture: an E(n)-equivariant GNN with a two-branch design that processes interface edges and internal edges separately, then pools with global attention and predicts pDockQ. This keeps rotational/translational symmetry while learning local interface geometry.
9. On the hardest docking benchmark setting (unbound–unbound docking; n=215), DeepRank-Ab variants outperform HADDOCK EMscoring and VoroIF-jury in Top-k success. Atom-distance edges do best at Top1, while Voronoi-area edges become strongest at higher k (e.g., Top10), reflecting tradeoffs between sharp early ranking and broader enrichment.
10. Generalization beyond docking decoys: on an AF3-focused external test set (59 complexes released after AF3’s training cutoff), DeepRank-Ab improves AF3 Top1 success by 35.5% and more than doubles mean Top1 DockQ. A notable practical tweak is removing the electrostatics term to better handle unrelaxed AF3 structures (distribution shift from clashes/packing), boosting Top1 success to 54.24%.
11. External CAPRI/MassiveFold evaluation (5 antibody–antigen targets) suggests strong out-of-distribution performance: DeepRank-Ab reaches 100% Top5 success, with especially strong results on peptide antigens. The paper also analyzes why: peptide interfaces tend to be more locally driven and compatible with the model’s 5 Å heavy-atom cutoff graph construction.
12. The work argues that “consensus scoring” is not automatically beneficial: combining DeepRank-Ab with AF3 via averaging or jury voting reduces performance, implying DeepRank-Ab already captures much of the useful ranking signal for these systems.
💻Code: github.com/haddocking/DeepRa…
📜Paper: doi.org/10.1038/s42003-026-1…
#ComputationalBiology #StructuralBioinformatics #Antibodies #ProteinDocking #GeometricDeepLearning #GNN #AlphaFold3 #HADDOCK #CAPRI #BenchmarkDatasets
8
49
3,005
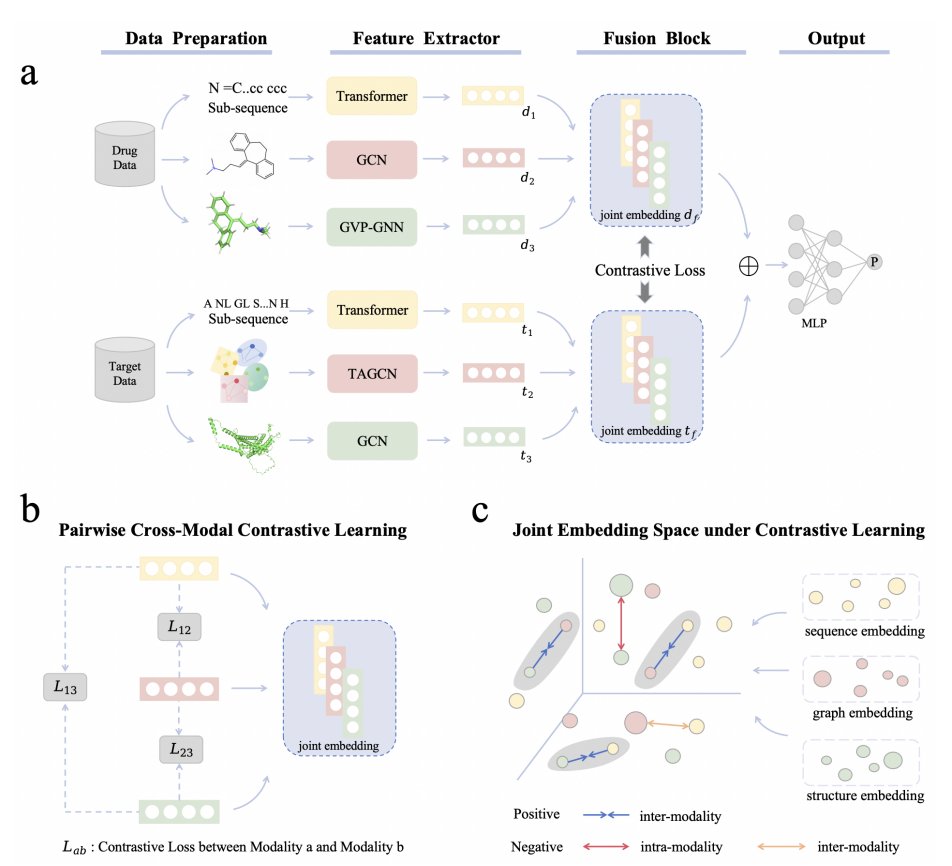
A Triple-Modal Contrastive Learning Framework with Sequence, Graph, and 3D Features for Drug–Target Interaction Prediction
1 TriMod-DTI is presented as a triple-modal DTI framework that jointly models 1D sequences, 2D graphs, and 3D structures for both drugs and proteins, aiming to learn universal yet complementary representations rather than relying on single- or bi-modal fusion.
2 The paper motivates tri-modality with an embedding cosine-similarity analysis (on GPCR) showing low similarity across 1D/2D/3D embeddings (mostly within [-0.25, 0.25]), suggesting strong complementarity and that explicit cross-modal modeling is needed.
3 For drugs, TriMod-DTI encodes: (i) SMILES sequences segmented by FCS and processed by a Transformer; (ii) 2D molecular graphs from RDKit with atom features (75-d) encoded by a GCN; (iii) 3D molecular graphs built from SDF coordinates with edges by distance (<4.5 Å) encoded by a GVP-GNN to integrate scalar/vector geometric features.
4 For proteins, TriMod-DTI encodes: (i) amino-acid sequences (FCS Transformer); (ii) binding-site pocket graphs extracted from OmegaFold-predicted structures, pocket detection via prior method, then TAGCN attention pooling to get pocket-aware embeddings; (iii) a residue-level 3D structural graph (Cα nodes; edges via 8 Å neighbor search) encoded with a GCN.
5 A core methodological piece is triple-modal cross-modal contrastive learning (inspired by CLIP-style alignment): embeddings of the same entity (drug or protein) across modalities form positive pairs (1D–2D, 2D–3D, 1D–3D), while other entities in-batch form negatives, aligning modalities to reduce distribution mismatch before fusion.
6 After alignment, the model concatenates all six embeddings (d1⊕d2⊕d3⊕t1⊕t2⊕t3) and uses an MLP classifier for binary DTI prediction; the total objective combines cross-entropy with separate contrastive losses for drugs and targets weighted by hyperparameters.
7 On three benchmarks (Human, GPCR, DrugBank), TriMod-DTI reports consistent improvements in AUC and often AUPR/Precision versus baselines spanning sequence-only and sequence graph methods; notably on GPCR it improves AUPR and Precision over a strong multi-attention baseline, while on DrugBank it yields best AUC/Precision but lower AUPR, attributed to class imbalance.
8 Ablations indicate the full tri-modal contrastive objective matters: removing contrastive learning or any cross-modal component degrades performance; the full contrastive setup is reported to improve over a non-contrastive variant (e.g., 1.1% AUC and 2.0% AUPR in their summary).
9 Modality-only analysis suggests sequence contributes most, graph next, and 3D alone is weakest in their setup; the authors argue 3D still adds complementary local spatial context when combined, and note a limitation that their 3D drug encoder may omit key chemical attributes (e.g., atom types/charges), leaving room for improved geometric/chemical featurization.
10 A case study ranks candidate targets for Verapamil and reports literature support for 5 of the top-10 predictions; docking for a top-ranked predicted target (Glucose-6-phosphate isomerase 2) suggests plausible hydrogen-bonding interactions in the pocket, illustrating potential utility for hypothesis generation.
💻Code: github.com/klez1/TriMod-DTI
📜Paper: arxiv.org/abs/2605.29926
#DrugDiscovery #DTI #MachineLearning #DeepLearning #MultimodalLearning #ContrastiveLearning #GeometricDeepLearning #Cheminformatics #Bioinformatics #ComputationalBiology
9
974
Geometric Flow Matching for Molecular Conformation Generation via Manifold Decomposition
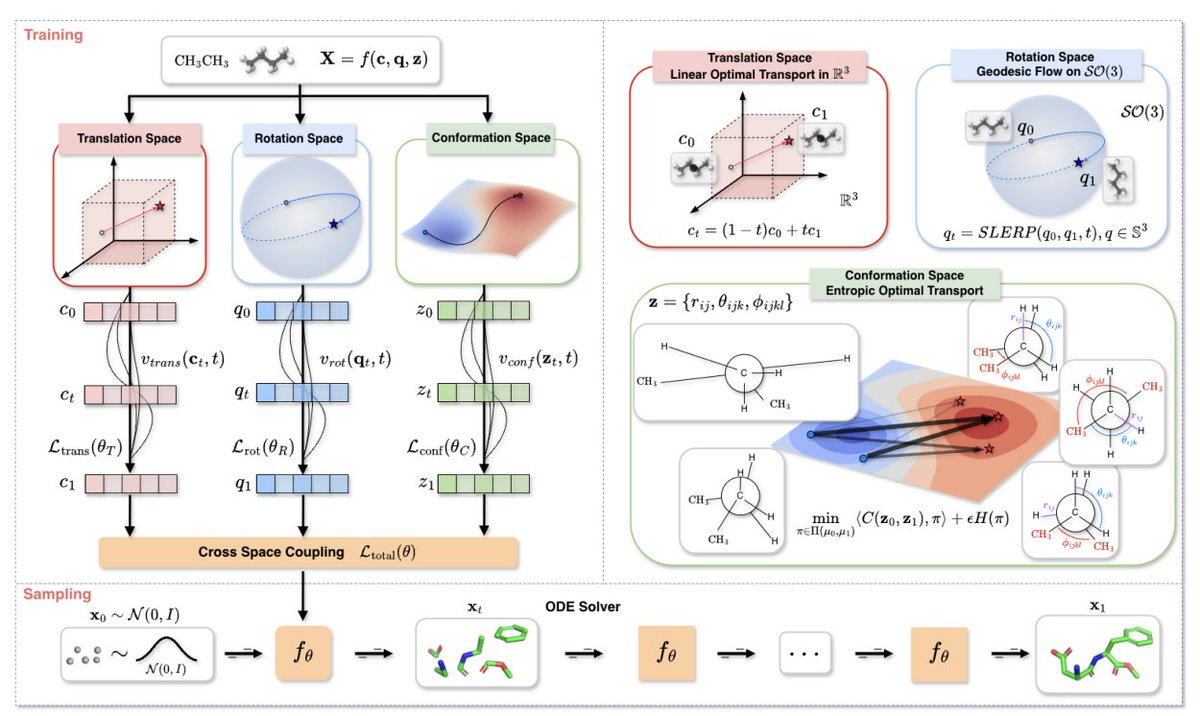
1. GO-Flow reframes molecular conformer generation as motion on the right manifolds rather than Cartesian point-cloud denoising, explicitly separating stiff vs. flexible degrees of freedom (rigid bond lengths/angles vs. flexible torsions) to avoid physically implausible intermediate structures.
2. The core idea is a manifold decomposition of a 3D molecule into three coupled subspaces: translation MT = R3 (center of mass), rotation MR = SO(3) (global orientation), and conformation MC (internal coordinates: bond lengths in R , bond angles in (0, π), torsions on a torus).
3. Each subspace uses a geometry-matched flow: translation uses linear optimal transport with constant velocity; rotation uses geodesic flows on SO(3) via unit quaternions and SLERP (avoiding Euler-angle singularities); conformation uses entropic optimal transport to traverse a multimodal internal-coordinate landscape without cutting through high-energy regions.
4. For rotation, GO-Flow trains on Lie-algebra angular velocities derived from relative quaternions (via log map), ensuring the learned velocity corresponds to a valid axis-angle rotation and remains consistent with rotation-equivariant neural architectures.
5. For internal coordinates, GO-Flow computes an entropic OT coupling (Sinkhorn) between noise and data internal-coordinate distributions, then defines the target velocity by barycentric projection of the OT plan; this is intended to better preserve diverse conformational modes while maintaining chemical plausibility.
6. The method composes the three learned velocities back into Cartesian atomic velocities using a Jacobian-based projection from internal coordinates to Cartesian space, yielding a total update that adds: translation, rotational cross-product motion around the center of mass, and J(z) vconf for internal-coordinate-driven motion.
7. Training uses a stability-oriented curriculum: (i) learn each manifold flow separately, (ii) jointly couple them with a Cartesian flow-consistency loss and uncertainty-based loss weighting, and (iii) optionally fine-tune as a continuous normalizing flow with likelihood evaluation using a Hutchinson trace estimator.
8. On GEOM-Drugs and GEOM-QM9, GO-Flow reports state-of-the-art coverage/matching tradeoffs while sampling with only 50 ODE solver steps; on GEOM-Drugs it achieves COV-R 94.82% and MAT-R 0.797 Å at 50 steps, compared with GeoDiff needing 5,000 steps for lower recall and worse matching.
9. Ablations indicate the conformation manifold (entropic OT over internal coordinates) contributes the largest gains; removing it causes the biggest drop in both recall and matching, while removing SO(3) rotation handling also noticeably degrades performance, supporting the claim that respecting manifold structure improves both validity and efficiency.
📜Paper: arxiv.org/abs/2605.25577
#ComputationalChemistry #MolecularGeneration #DiffusionModels #FlowMatching #OptimalTransport #SO3 #EquivariantML #DrugDiscovery #GeometricDeepLearning #ConformerGeneration
5
26
1,328

We’re excited to welcome Petar Veličković to the OxML speaker lineup.
Petar is a Senior Staff Research Scientist at Google DeepMind, and a Lecturer at University of Cambridge. His research focuses on neural algorithmic reasoning, graph representation learning, and geometric deep learning, with the goal of improving out-of-distribution generalisation in AI systems.
He is widely known as the first author of Graph Attention Networks (GAT) and Deep Graph Infomax, with research that has impacted areas ranging from Google Maps travel-time prediction to mathematical discovery, football tactics, and competitive programming.
Join us to hear insights from one of the leading researchers shaping the future of graph and geometric deep learning.
Register Now: oxfordml.school
#OxML #GraphAttentionNetworks #GraphRepresentationLearning #RepLearning #GeometricDeepLearning @PetarV_93 @GoogleDeepMind @Cambridge_Uni

3
106

💻 Code: github.com/PedrV/gnn-uq-insp…
📄 Preprint: arxiv.org/abs/2605.22593
Co-authored w/ @PedroCVieira0 & @pedroribeiro_pt.
#MachineLearning #GraphNeuralNetworks #DeepLearning #UncertaintyQuantification #GeometricDeepLearning
1
3
87
ConTact: Contact-First Antibody CDR Design via Explicit Interface Reasoning
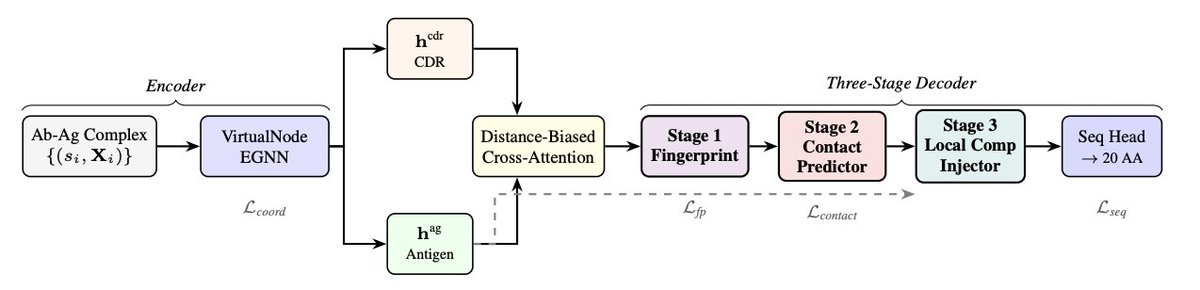
1. CONTACT reframes antigen-conditioned antibody CDR design as two distinct problems that should not be conflated: (a) deciding which CDR positions will actually contact the antigen (the “where”), and (b) choosing amino acids at those positions (the “what”). The paper argues current models often underuse antigen information because they try to solve both implicitly with uniform message passing and uniform sequence loss.
2. The core architectural idea is a contact-then-act, three-stage cascade for CDR-H3: Stage 1 learns per-position “surface complementarity fingerprints”; Stage 2 explicitly predicts which CDR residues contact the antigen (supervised); Stage 3 injects antigen features into the sequence head only where contacts are predicted, so antigen signal is routed preferentially to binding-critical positions.
3. Stage 1 (Complementarity fingerprinting) produces a compact vector per CDR position summarizing the local binding environment, inspired by molecular surface fingerprints. It is trained with an InfoNCE-style contrastive objective so positions facing similar antigen environments have similar fingerprints, improving downstream contact prediction.
4. Stage 2 (Contact prediction) uses a supervised contact label defined by a Cα–Cα threshold of 8 Å. The predictor combines CDR embeddings, KNN-aggregated antigen features, minimum-distance encodings, and the Stage 1 fingerprint. A focal binary cross-entropy loss addresses contact/non-contact imbalance and focuses learning on ambiguous boundary cases.
5. Stage 3 (Contact-guided injection) performs “double gating” to control antigen-conditioning strength at each CDR position: a learned gate multiplied by the predicted contact probability. This aims to prevent distant/noisy antigen residues from influencing non-contact positions, while still allowing fine-grained modulation at predicted contact sites.
6. The model also adds a distance-biased cross-attention module: standard cross-attention scores are augmented with a Gaussian bias based on predicted Cα distances, encoding a geometric prior that spatial neighbors should matter more for binding than far-away residues.
7. On the encoder side, CONTACT uses a heterogeneous VirtualNode-EGNN with virtual nodes connecting to all epitope residues and all CDR residues, creating a two-hop shortcut for epitope-to-CDR information flow and mitigating over-squashing that can occur when signals must traverse long chains of message passing steps.
8. Training uses a multi-term objective centered on sequence loss plus explicit contact loss and fingerprint loss, along with coordinate, pairing (CDR–antigen matching), docking (encouraging proximity to the epitope), and auxiliary regularization terms. A key detail is contact-weighted cross-entropy for sequence prediction: positions with higher predicted contact probability receive larger weights, concentrating gradient on binding-relevant residues.
9. Results on CHIMERA-BENCH (2,922 complexes; epitope-group split) show CONTACT leading on structural and interface awareness metrics among 11 retrained baselines: RMSD 1.63 Å (7% better than next-best), epitope F1 0.79 (10% over GNN baselines), fnat 0.67, DockQ 0.73, and competitive sequence recovery AAR 0.38. The paper highlights that CAAR remains low (0.20) across methods, suggesting a remaining bottleneck: Cα-level antigen representations may not capture enough chemistry (side chains/electrostatics) to nail residue identity at contacts.
📜Paper: arxiv.org/abs/2605.21600
#ComputationalBiology #AntibodyDesign #ProteinDesign #GeometricDeepLearning #GNN #EquivariantNetworks #StructuralBiology #MachineLearning
3
12
1,796
EvoStruct: Bridging Evolutionary and Structural Priors for Antibody CDR Design via Protein Language Model Adaptation
1. EvoStruct addresses a practical failure in structure-conditioned antibody CDR design: equivariant GNNs can score high on sequence recovery but collapse their outputs to a tiny amino-acid vocabulary (often overpredicting Tyr/Gly and missing rare but important residues like Trp/Cys/Met).
2. The paper traces vocabulary collapse to a root cause: GNN encoders must learn amino-acid distributions de novo from limited structural complexes (~3k), effectively discarding evolutionary substitution patterns that protein language models (PLMs) already encode from hundreds of millions of sequences.
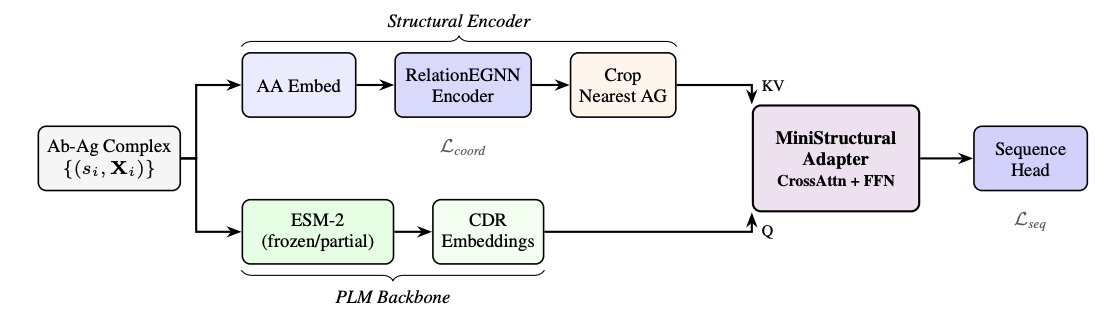
3. Core idea: keep sequence prediction in a PLM’s calibrated embedding space while injecting 3D context. EvoStruct bridges frozen/partially-frozen ESM-2 with an E(3)-equivariant relation-aware GNN using a cross-attention “mini structural adapter” operating in ESM-2 representation space.
4. Architectural separation of concerns: the GNN focuses on geometry of the antibody–antigen complex (typed edges, equivariant coordinate updates), while ESM-2 provides evolutionary priors and well-calibrated amino-acid probabilities; cross-attention lets structural context modulate PLM representations without overwriting them.
5. Training specifically targets CDR design pathologies (not just generic inverse folding): progressive PLM unfreezing to avoid catastrophic forgetting of evolutionary priors, plus R-Drop consistency regularization to stabilize predictions under dropout and encourage robust context injection.
6. The study introduces/uses diagnostics to quantify collapse and conditioning: effective vocabulary (Veff, exponentiated entropy) and paratope–epitope amino-acid pair correlation, alongside standard metrics like AAR/CAAR, perplexity, RMSD, fnat, DockQ, epitope F1, and sequence liabilities.
7. On CHIMERA-BENCH (epitope-group split; 2,922 SAbDab complexes), EvoStruct ranks best on sequence metrics: AAR 0.43 (vs 0.37 best GNN baselines; 16% relative) and perplexity 1.88 (vs 3.27 RAAD; -43%), while maintaining comparable backbone RMSD (~1.84 Å).
8. EvoStruct substantially reduces vocabulary collapse: Veff = 12.4 (~80% of native diversity) vs GNN greedy-decoding baselines Veff ~4.7–5.3 (~30–34% of native). It also recovers far more motifs (e.g., 282 unique bigrams and 1,214 trigrams vs RAAD 52/110), indicating improved diversity without sacrificing accuracy.
9. Conditioning improves where baselines struggle: EvoStruct shows the strongest binding-pair learning (paratope–epitope pair correlation r = 0.73; interface enrichment correlation r = 0.81), suggesting cross-attention successfully routes antigen information to the sequence head, though contact-position recovery remains difficult for all methods.
10. Limitations noted: EvoStruct does not lead binding-quality metrics (e.g., RefineGNN—without antigen conditioning—still tops fnat/DockQ), and contact AAR remains low (~22%), implying future work may need richer antigen-conditioning mechanisms or objectives tailored to interface positions.
📜Paper: arxiv.org/abs/2605.21485
#ComputationalBiology #AntibodyDesign #ProteinDesign #ProteinLanguageModels #GeometricDeepLearning #GNN #ESM2 #InverseFolding #CDR #MachineLearning
4
15
1,876
Hierarchical Contrastive Learning for Multi-Domain Protein-Ligand Binding
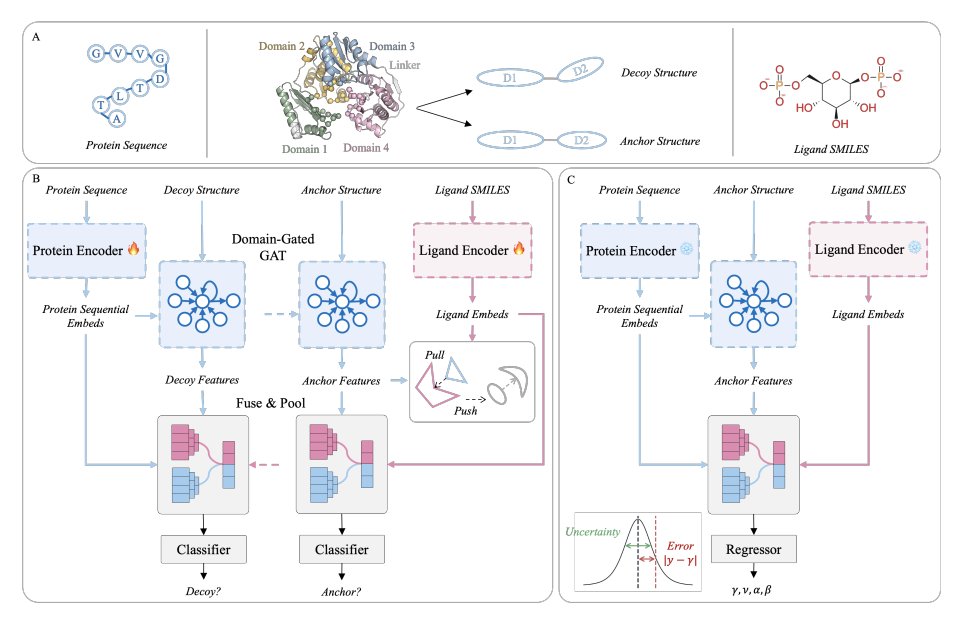
1. HCLBind reframes protein–ligand affinity prediction for multi-domain targets as a hierarchical geometry problem: instead of treating proteins as monolithic rigid graphs, it explicitly learns both local pocket rules and global inter-domain conformational validity, which is critical when domain motions control access and recognition.
2. The key idea is to decouple representation learning from affinity regression via self-supervised pre-training on Q-BioLiP, then fine-tune on PDBbind. This reduces reliance on scarce labeled affinity data and makes the learned features more robust to flexible regions and misleading geometries.
3. A novel hierarchical decoy strategy provides structure-aware supervision at two levels: for single-domain proteins it perturbs coordinates with Gaussian noise (σ=1.5 Å) to teach local physicochemical/geometric constraints; for multi-domain complexes it rotates one domain (15°/30°) to create interface-invalid conformations that test global quaternary geometry.
4. Pre-training combines two complementary objectives: Interface Decoy Discrimination (IDD) as a binary task to separate native vs perturbed interfaces (learning “is this interface physically valid?”), and Ligand–Protein Matching (LPM) with an InfoNCE batch contrastive loss to learn physicochemical compatibility between proteins and ligands.
5. Architecturally, HCLBind uses a dual-branch multimodal encoder: ESMC for protein sequence embeddings and MolFormer for ligand SMILES embeddings, then a domain-gated graph attention network for protein structure where a learnable interface bias prioritizes inter-domain edges while masking non-neighbors.
6. Cross-modal fusion is performed with multi-head cross-attention where ligand tokens query protein structural tokens, approximating ligand “surface scanning” and encouraging the model to focus on discriminative interface features rather than diffuse global signals.
7. Parameter-efficient adaptation is done with LoRA on the protein and ligand foundation model backbones to preserve evolutionary/chemical knowledge while still adapting to binding tasks under limited supervision; ablations show removing LoRA substantially degrades accuracy.
8. For fine-tuning, HCLBind uses Evidential Deep Learning (EDL) with a Normal-Inverse-Gamma output to model both epistemic and aleatoric uncertainty, targeting the known issue that flexible linker regions inject noise and can cause overconfident predictions in deterministic regressors.
9. On a strict time-based PDBbind split (test complexes released ≥2019), HCLBind reports RMSE 1.309, PCC 0.698, C-index 0.744, outperforming multiple baselines including a strong contrastive baseline (CL-GNN). Ablations indicate IDD and LPM are both necessary, and EDL improves both performance and reliability.
10. Reliability analyses show uncertainty is actionable: rejecting high-uncertainty predictions monotonically reduces RMSE, and the model assigns significantly higher epistemic uncertainty to structurally disrupted interface decoys than to native complexes, suggesting it internalizes geometric validity signals from IDD.
💻Code: github.com/jiankliu/HCLBind
📜Paper: arxiv.org/abs/2605.19902
#ComputationalBiology #GeometricDeepLearning #DrugDiscovery #ProteinLigand #GNN #ContrastiveLearning #UncertaintyEstimation #ProteinDesign #MultimodalAI #MachineLearning
7
23
1,708

📣 LOGML'26 Speaker Series
🎤 Soledad Villar @SoledadVillar5 (Johns Hopkins)
Her research spans equivariant ML, GNNs, with applications to computational biology.
📍 Imperial College London
📅 13–17 July 2026
🔗 logml.ai/
#LOGML #GraphML #GeometricDeepLearning

3
10
408

May 19
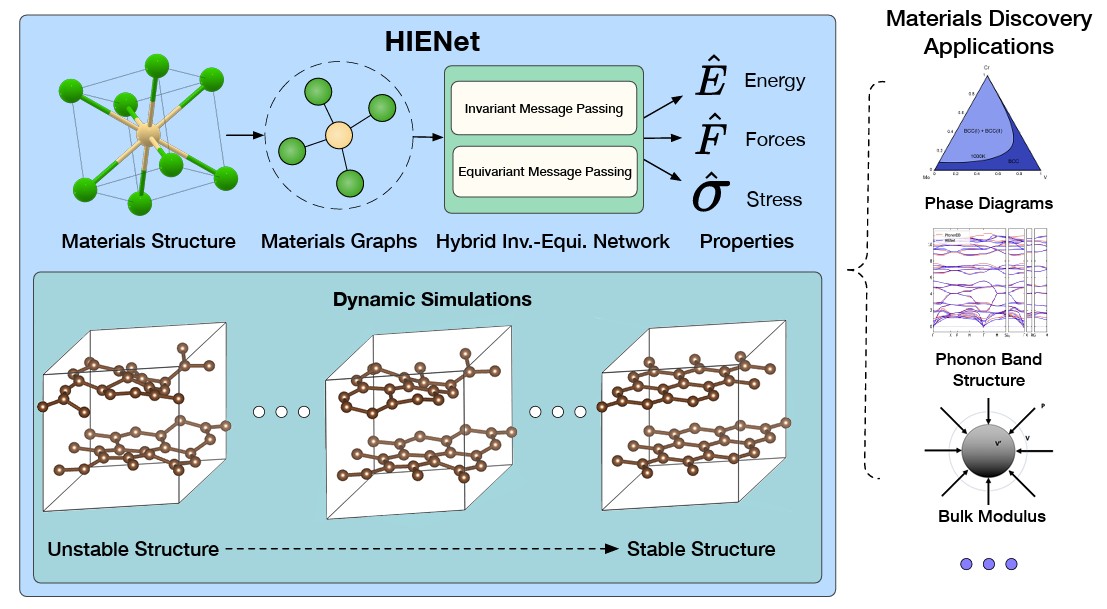
🚀 Excited to share our work on materials foundation models, which is accepetd by Transactions on Machine Learning Research @TmlrOrg @TmlrPub:
"Learning Materials Interatomic Potentials via Hybrid Invariant-Equivariant Architectures"
📄 Paper: openreview.net/forum?id=fq3n…
💻Code: github.com/divelab/AIRS/tree…
⚛️ Machine learning interatomic potentials (MLIPs) are critical for predicting energy, force, and stress in materials. It powers downstream tasks from materials discovery to molecular simulation.
But there is a key tradeoff:
⚡ Invariant models → efficient, scalable, but can struggle with higher-order physics
🧠 Equivariant models → physically expressive, symmetry-aware, but computationally expensive
💡 We introduce HIENet, a hybrid invariant–equivariant architecture that combines the best of both worlds:
✅ Integrates invariant equivariant message passing
✅ Provably satisfies key physical constraints
✅ Strong accuracy–efficiency tradeoff
✅ Significant computational speedups over prior methods
✅ Scales across model sizes & different equivariant backbones
Our experiments on benchmark datasets downstream materials discovery tasks show that hybrid architectures may be a powerful direction for next-generation MLIPs 🔬
Great collaboration with amazing researchers
@KeqiangY, Montgomery Bohde, Andrii Kryvenko, Ziyu Xiang, Kaiji Zhao, Siya Zhu, Saagar Kolachina, Doguhan Sarıtürk, Raymundo Arróyave, Xiaoning Qian, Xiaofeng Qian, @ShuiwangJi @TAMU
#AI4Science #MaterialsFoundationModel #MachineLearning #GeometricDeepLearning #FoundationModels #ForceFieldPrediction #ComputationalChemistry #TMLR #MolecularDynamics #icml #neurips #iclr
3
191
We are excited to have Professor Michael Bronstein, a leading figure in geometric deep learning, whose work has advanced machine learning on graphs and non-Euclidean domains, shaping modern AI across computer vision, representation learning, and large-scale real-world systems.
Michael Bronstein is the DeepMind Professor of AI at the University of Oxford and Founding Scientific Director, AI at the AITHYRA Research Institute for Biomedical Artificial Intelligence in Vienna.
His research interests lie in geometric deep learning, graph neural networks, 3D shape analysis, protein design, non-human species communication.
Limited Slots available!
Register for MLx Health & Bio: oxfordml.school
#OxML2026 #MLxHealth #MachineLearning #AIinHealthcare #GeometricDeepLearning @mmbronstein @UniofOxford

3
15
2,299
Steering semi-flexible molecular diffusion model for structure-based drug design with reinforcement learning @ScienceAdvances
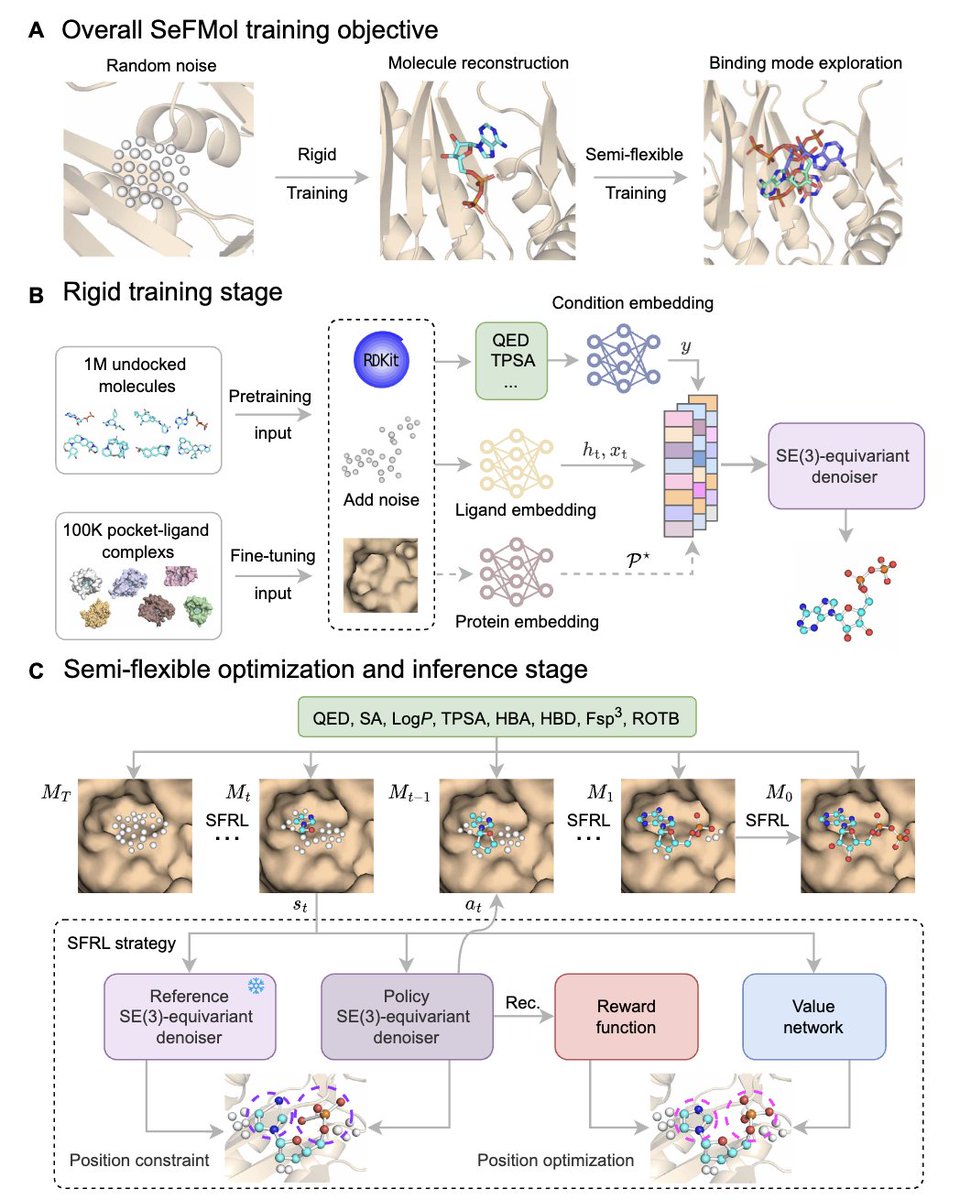
1. The paper introduces SeFMol, a reinforcement learning (RL)–steered 3D diffusion model that treats diffusion denoising as a Markov decision process, enabling semi-flexible, stepwise conformational adjustment of ligands inside protein pockets rather than assuming rigid ligand conformations.
2. Core idea: a pretrained “rigid” conditional diffusion model is further optimized with a semi-flexible RL stage (SFRL). A frozen reference denoiser anchors the policy denoiser via a KL constraint, helping prevent reward overfitting and catastrophic drift while improving binding-oriented geometry.
3. Property control is built into generation as explicit conditioning signals (computed with RDKit): QED, SA, LogP, TPSA, HBA, HBD, Fsp3, and ROTB. These conditions act as dense guidance throughout denoising to counter sparse terminal rewards from docking-based objectives.
4. Training pipeline: (i) pretrain on 1,000,000 target-free Molecule3D molecules with property conditioning to learn general structure–property priors; (ii) fine-tune on 100,000 CrossDocked2020 protein-ligand pairs to become pocket-aware while retaining property bias; (iii) apply SFRL to steer denoising trajectories toward better pocket complementarity.
5. Efficiency contribution: a variable fast sampling strategy reduces diffusion steps from 1000 (training) to 50 (sampling/optimization), yielding ~20x fewer steps while maintaining quality; reported sampling is ~0.81 s per molecule with 98.3% completion, substantially faster than several diffusion baselines.
6. Docking-centric results on 100 test pockets (100 molecules per pocket): SeFMol reports average Vina score −7.23 kcal/mol and improved affinity-related metrics (Vina min and Vina dock). It also reports an SR (success rate under nine joint affinity property constraints) of 11.53%.
7. Geometric/interaction reliability: SeFMol’s generated poses show strong agreement between direct Vina scoring and redocking (reported correlation 0.95), plus competitive RMSD-to-redocked distributions and fewer docking clashes, consistent with the goal of generating chemically plausible 3D conformations directly.
8. Interaction-pattern preservation with optimization: using PLIP interaction typing, SeFMol maintains interaction-type distributions close to reference ligands (reported JSD 0.1401, comparable to the best baseline), suggesting affinity gains do not come from unrealistic interaction artifacts.
9. Generalization case studies: on real-world targets CDK2 (1H00) and ROCK1 (6E9W), SeFMol-generated ligands reproduce canonical interactions seen in known actives while also proposing alternative chemotypes and plausible new interactions; property steering (e.g., setting TPSA or Fsp3 targets) shifts generated distributions around specified values.
📜Paper: doi.org/10.1126/sciadv.ady99…
#ComputationalBiology #StructureBasedDrugDesign #DiffusionModels #ReinforcementLearning #MolecularGeneration #GenerativeAI #DrugDiscovery #GeometricDeepLearning #SE3Equivariance
1
5
15
1,805

Apr 18
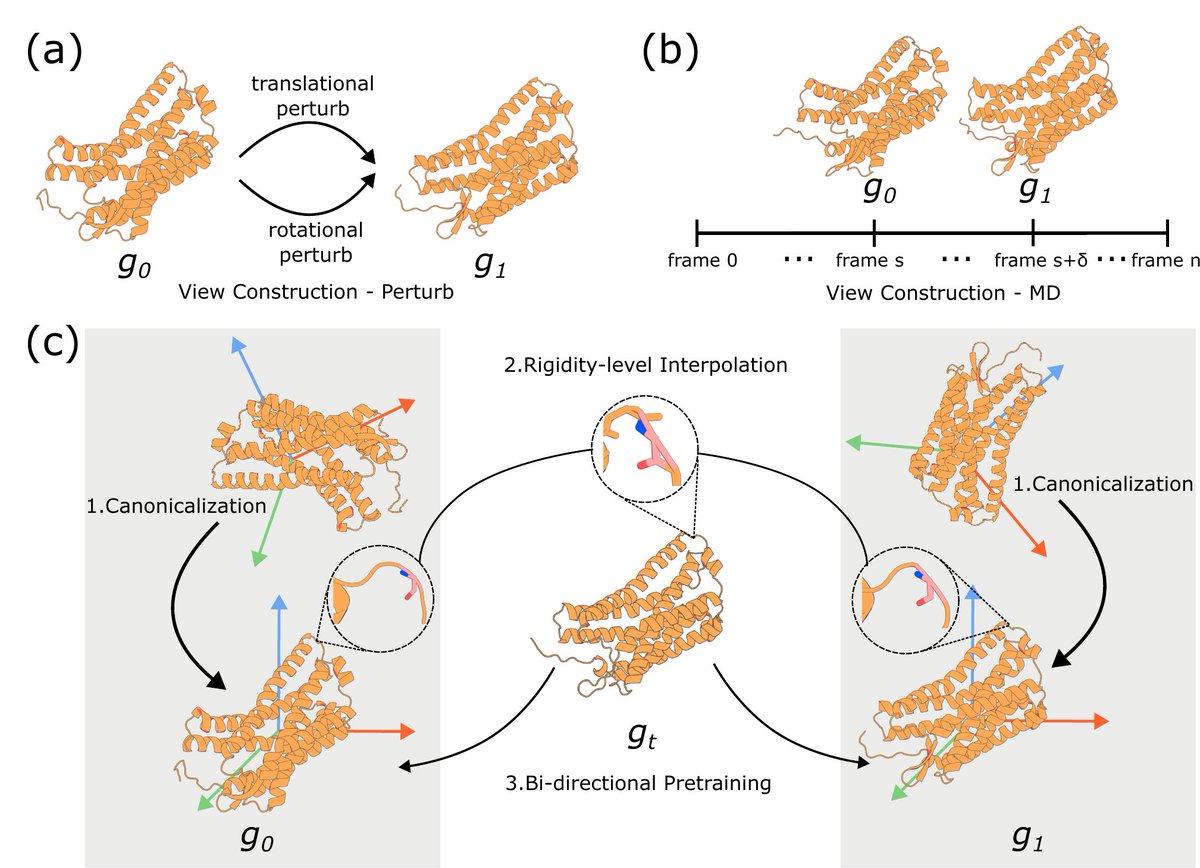
(1/3) Excited to share our work RigidSSL — “Rigidity-Aware Geometric Pretraining for Protein Design & Conformational Ensembles” — accepted at #ICLR2026 🇧🇷🪨
Most protein generative models try to learn geometry generation simultaneously.
That’s a tough optimization problem — and likely suboptimal.
🪨 RigidSSL: decouple the two.
We first pretrain geometric priors, then finetune for generation:
→ large-scale AlphaFold structures
→ molecular dynamics trajectories
Simple idea. Surprisingly effective.
Results:
• 43% improvement in designability
• More realistic GPCR conformational ensembles
• Best on 7/9 evaluation metrics
Pretrain geometry. Then generate.
Sometimes, structure should come before creativity.
#ICLR #AI4Bio #ProteinDesign #GenerativeModels #GeometricDeepLearning #GenAI
1
7
59
10,523

Apr 15
University of Bergen | PhD in Geometry & Machine Learning 🤖📐
🚨 Deadline: April 30, 2026
Join University of Bergen 🇳🇴 for an exciting PhD at the intersection of mathematics, geometry, and machine learning.
📌 Position: PhD Research Fellow – Geometry for Machine Learning
🏫 Department: Mathematics
📍 Location: Bergen, Norway
👨🏫 Supervisor: Prof. Erlend Grong
💰 Salary: NOK 568,700/year
📆 Duration: 3 years (with possible 4th year including teaching)
💡 Project Focus
Work on Geometric Deep Learning (GDL)—a cutting-edge area where geometry and symmetry enhance machine learning models beyond traditional approaches 📐
🔬 What You’ll Work On
• 🤖 Develop equivariant neural networks
• 📊 Apply ML to data with geometric structures (graphs, surfaces, manifolds)
• 🧮 Explore theory: group theory, differential geometry & representation theory
• ⚙️ Implement models, run experiments & evaluate performance
• 🌐 Solve problems involving PDEs, transfer learning & stochastic processes
🌍 Research Impact
• Advance next-gen AI models grounded in mathematics
• Improve learning on complex structured data
• Contribute to both theoretical math & real-world ML applications
• Be part of the rapidly growing Geometric Deep Learning field
🎯 Ideal Candidate
• Master’s in Mathematics or CS (strong math background)
• Knowledge of machine learning group theory basics
• Interest in geometry, manifolds & advanced math concepts
• Experience training ML models
• Strong analytical & collaboration skills
🎁 Why Apply?
• 💰 Competitive salary public pension benefits
• 🌍 Work in a strong academic environment in Norway
• 📚 Structured PhD training teaching opportunities
• 🤝 Collaborate across mathematics & informatics departments
• 📈 Excellent career growth in AI & academia
🌆 Why Bergen?
A stunning coastal city known for its fjords, nature, and high quality of life 🌄
🔗 More Info
phdscanner.com/opportunities…
🚨 Apply before April 30, 2026!
#PhD #MachineLearning #ArtificialIntelligence #Mathematics #GeometricDeepLearning #Norway #UniversityOfBergen #ResearchJobs #PhDOpportunity
2
13
497
Interpretable Antibody–Antigen Structural Interface Prediction via Adaptive Graph Learning and Cyclic Transfer
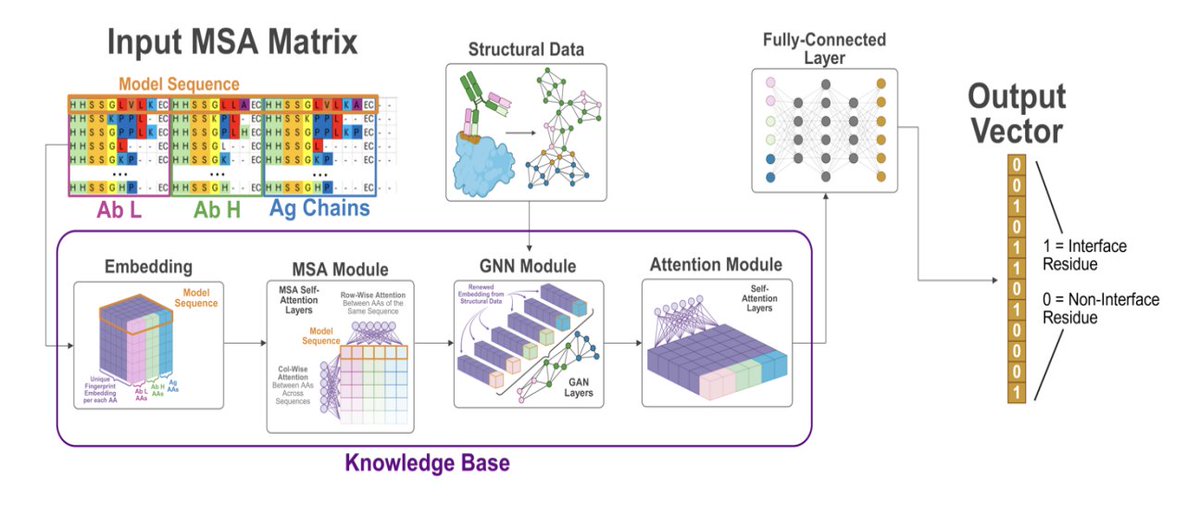
1. The paper introduces VASCIF (Variable-domain Antibody–antigen Structural Complex Interface Finder), a structure-aware model that jointly predicts paratopes and epitopes at residue resolution from antibody–antigen complex structures, explicitly targeting the extreme class imbalance where only ~5–10% of residues are interface residues.
2. Core modeling idea: complexes are represented as residue graphs (nodes=residues; edges=spatial proximity), and a Masked Graph Attention (MGA) architecture uses attention-based message passing to capture non-local, long-range structural dependencies that matter for binding.
3. A key innovation is Dynamic Masking (DyM): a learnable gating module that suppresses low-information residues and amplifies contextually informative ones end-to-end, avoiding common heuristics like CDR-only restriction or negative downsampling; ablations show removing DyM notably hurts epitope AUPR (e.g., 0.472 → 0.458 on Paragraph).
4. Another innovation is Cyclic Transfer with Soft Restart (CTSR): a lightweight cyclic training strategy that alternates interface prediction with auxiliary structural tasks (secondary structure and contact map prediction). This temporal cycling (rather than simultaneous multitask learning) is designed for small structural datasets and helps optimization/generalization; it improves AUPR for both sides (e.g., Paragraph epitope AUPR 0.472 → 0.490; paratope 0.765 → 0.778).
5. The work emphasizes that AUROC can be misleading under heavy imbalance, so it optimizes and reports AUPR as the primary metric, using an imbalance-aware objective that combines class-weighted cross-entropy, a differentiable AUPR surrogate, and a graph smoothness regularizer encouraging consistent predictions for neighboring residues.
6. Benchmarking spans three settings with leakage-aware evaluation: Paragraph-expanded (1,086 complexes; 5-fold CV), MIPE (626 complexes; standard split with CV), and VASCO (virus-focused; cluster-based splitting to reduce sequence leakage). Across datasets, VASCIF is consistently the best-performing method among models that predict both antibody and antigen interfaces.
7. A notable biophysical/labeling insight: redefining “interface” using a larger residue distance threshold (~10 Å) rather than the conventional 4.5 Å improves predictive performance substantially (e.g., on VASCO with CTSR and 10 Å: paratope AUPR 0.882 and epitope AUPR 0.663), supporting the view of interfaces as interaction neighborhoods influenced by longer-range forces.
8. Interpretability results: DyM mask profiles preferentially highlight flexible loop regions and down-weight well-packed helices/sheets, aligning with known interface determinants (e.g., CDR loop involvement). The paper argues DyM provides more reliable residue-importance signals than last-layer attention maps, which can appear diffuse.
9. The learned interaction preferences are consistent with known Ab–Ag chemistry: antibody-side tyrosine/serine (and other aromatics) frequently contribute to binding hotspots; antigen-side enrichment of polar residues (e.g., asparagine/glutamine) is recovered in predicted interaction patterns, suggesting the model internalizes biophysically meaningful trends.
10. Practical takeaway: antibody-only paratope prediction is already strong (Paragraph AUPR 0.751), but adding antigen context measurably refines predictions (0.765; and 0.778 with CTSR), implying fast antibody-only screening is feasible while full complex modeling is preferable when antigen structure is available.
💻Code: github.com/SimBioSys-lab/MGA
📜Paper: biorxiv.org/content/10.64898…
#computationalbiology #bioinformatics #deeplearning #geometricdeeplearning #GNN #structuralbiology #antibodies #immunology #epitope #paratope #proteinproteininteraction #interpretability #machinelearning

3
31
7,605
Interpretable Antibody–Antigen Structural Interface Prediction via Adaptive Graph Learning and Cyclic Transfer
1. The paper introduces VASCIF (Variable-domain Antibody–antigen Structural Complex Interface Finder), a structure-aware model that jointly predicts paratopes and epitopes at residue resolution from antibody–antigen complex structures, explicitly targeting the extreme class imbalance where only ~5–10% of residues are interface residues.
2. Core modeling idea: complexes are represented as residue graphs (nodes=residues; edges=spatial proximity), and a Masked Graph Attention (MGA) architecture uses attention-based message passing to capture non-local, long-range structural dependencies that matter for binding.
3. A key innovation is Dynamic Masking (DyM): a learnable gating module that suppresses low-information residues and amplifies contextually informative ones end-to-end, avoiding common heuristics like CDR-only restriction or negative downsampling; ablations show removing DyM notably hurts epitope AUPR (e.g., 0.472 → 0.458 on Paragraph).
4. Another innovation is Cyclic Transfer with Soft Restart (CTSR): a lightweight cyclic training strategy that alternates interface prediction with auxiliary structural tasks (secondary structure and contact map prediction). This temporal cycling (rather than simultaneous multitask learning) is designed for small structural datasets and helps optimization/generalization; it improves AUPR for both sides (e.g., Paragraph epitope AUPR 0.472 → 0.490; paratope 0.765 → 0.778).
5. The work emphasizes that AUROC can be misleading under heavy imbalance, so it optimizes and reports AUPR as the primary metric, using an imbalance-aware objective that combines class-weighted cross-entropy, a differentiable AUPR surrogate, and a graph smoothness regularizer encouraging consistent predictions for neighboring residues.
6. Benchmarking spans three settings with leakage-aware evaluation: Paragraph-expanded (1,086 complexes; 5-fold CV), MIPE (626 complexes; standard split with CV), and VASCO (virus-focused; cluster-based splitting to reduce sequence leakage). Across datasets, VASCIF is consistently the best-performing method among models that predict both antibody and antigen interfaces.
7. A notable biophysical/labeling insight: redefining “interface” using a larger residue distance threshold (~10 Å) rather than the conventional 4.5 Å improves predictive performance substantially (e.g., on VASCO with CTSR and 10 Å: paratope AUPR 0.882 and epitope AUPR 0.663), supporting the view of interfaces as interaction neighborhoods influenced by longer-range forces.
8. Interpretability results: DyM mask profiles preferentially highlight flexible loop regions and down-weight well-packed helices/sheets, aligning with known interface determinants (e.g., CDR loop involvement). The paper argues DyM provides more reliable residue-importance signals than last-layer attention maps, which can appear diffuse.
9. The learned interaction preferences are consistent with known Ab–Ag chemistry: antibody-side tyrosine/serine (and other aromatics) frequently contribute to binding hotspots; antigen-side enrichment of polar residues (e.g., asparagine/glutamine) is recovered in predicted interaction patterns, suggesting the model internalizes biophysically meaningful trends.
10. Practical takeaway: antibody-only paratope prediction is already strong (Paragraph AUPR 0.751), but adding antigen context measurably refines predictions (0.765; and 0.778 with CTSR), implying fast antibody-only screening is feasible while full complex modeling is preferable when antigen structure is available.
💻Code: github.com/SimBioSys-lab/MGA
📜Paper: biorxiv.org/content/10.64898…
#computationalbiology #bioinformatics #deeplearning #geometricdeeplearning #GNN #structuralbiology #antibodies #immunology #epitope #paratope #proteinproteininteraction #interpretability #machinelearning
2
14
1,353
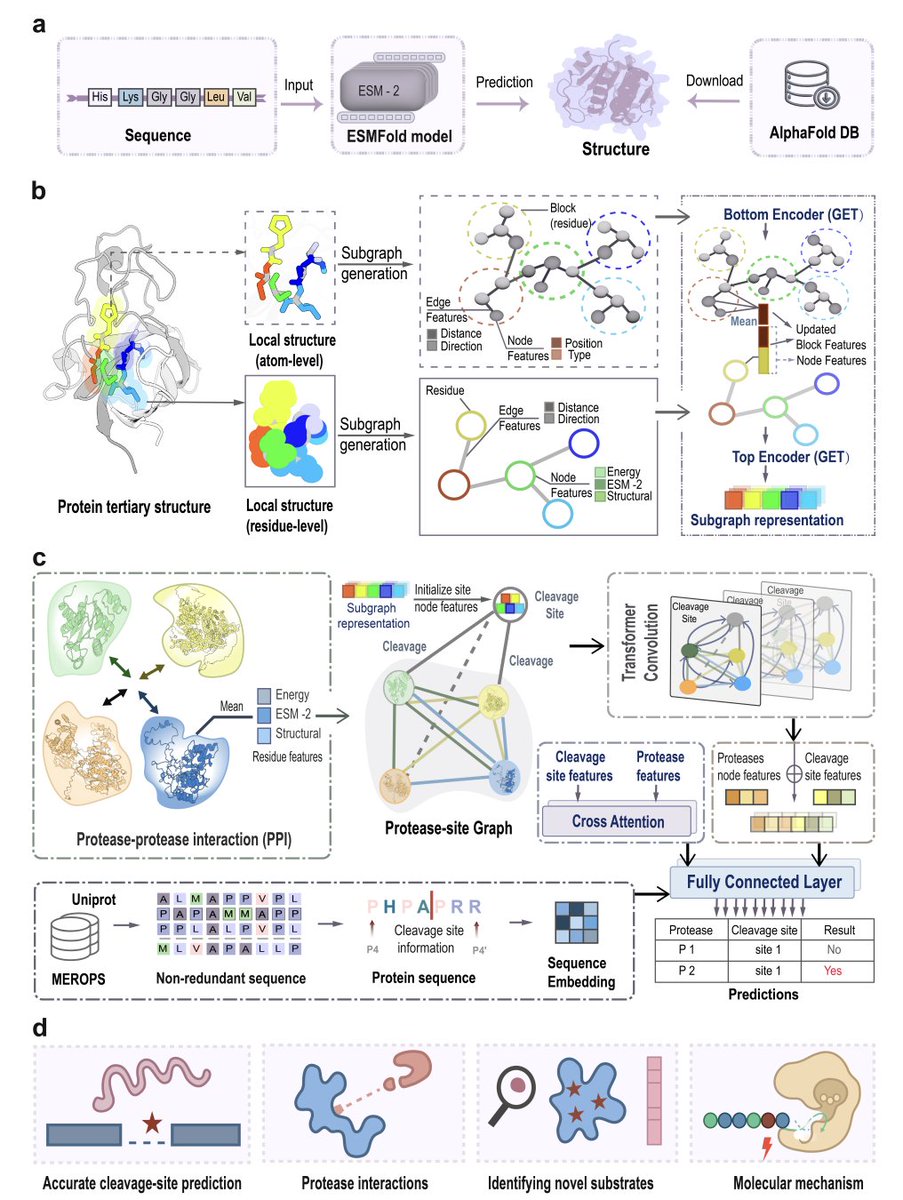
Structure-aware geometric graph learning for modeling protease–substrate specificity at scale
1. The paper introduces OmniCleave, a unified structure-aware geometric graph learning framework that predicts protease cleavage sites across 103 proteases (6 superfamilies) in a single model, aiming to move beyond motif-centric predictors by explicitly modeling 3D context and inter-protease dependencies.
2. Key idea: represent each candidate cleavage site by a cleavage-centric hierarchical structural graph built from predicted substrate structures (AlphaFold DB ESMFold). A residue-level subgraph captures residues within 10 Å of P1; an atom-level subgraph refines local chemistry/geometry. Edges encode distances (RBF) and directions (vectors), enabling geometric reasoning.
3. OmniCleave couples local substrate structure with a protease–protease interaction (PPI) module (STRING-derived). Proteases are nodes in a PPI graph; message passing provides relational priors so the model can learn cooperative or correlated cleavage behavior rather than treating proteases as isolated predictors.
4. Architecture: a hierarchical equivariant graph encoder (GET) updates atom- and residue-level representations, then a heterogeneous graph transformer links proteases to cleavage-site nodes, turning cleavage prediction into a protease–site link prediction problem that naturally supports many-to-one settings.
5. Scale of training data: 57,278 structure-informed protease–substrate cleavage events from MEROPS/UniProt, covering 9,651 substrates; negatives are balanced 1:1 with randomly sampled non-cleavage sites. Substrates are filtered for redundancy (CD-HIT; >70% identity removed), and training/test split is 7:3.
6. Benchmarking against six tools (Procleave, PROSPERous, DeepCleave, SitePrediction, PeptideCutter, ProsperousPlus) shows consistent gains. Reported results include AUC >0.9 for 48 proteases and >0.8 for 75 proteases; under a stricter similarity threshold (<30%), AUC >0.9 for 58 proteases and >0.8 for 74 proteases.
7. Many-to-one cleavage is treated as a first-class problem: MEROPS indicates some sites are cleaved by up to 20 proteases. On a many-to-one test subset, OmniCleave maintains high coverage even when ≥5 proteases target the same site, outperforming alternatives whose performance drops substantially—consistent with the benefit of PPI-informed relational learning.
8. Comparison with AlphaFold3 complex prediction (used as a proxy interface-based heuristic) suggests interface proximity alone misses many annotated sites. In case studies (Cathepsin L/E with P01317; MMP7 with P02671), OmniCleave recovers all annotated cleavage sites while AlphaFold3 identifies only a small subset.
9. Mechanistic interpretability: predictions mirror observed secondary-structure preferences of P1 residues across 54 human proteases (cleavages enriched in loops, α-helices, β-sheets, turns, bends). Feature perturbation highlights strong contributions from Rosetta energy terms (e.g., van der Waals/backbone constraints) and secondary-structure descriptors, supporting a geometry/energetics-driven view of specificity.
10. Experimental validation: in vitro Caspase-3 assays confirm 3 novel substrates (CUL7, THOC5, RPIA) and 21 cleavage sites detected by LC-MS/MS. OmniCleave correctly predicts 8/12 (THOC5), 5/6 (RPIA), and 8/12 (CUL7) sites, while Procleave predicts 0/12, 2/6, and 2/12, respectively; docking analyses provide plausible interaction rationales at example sites.
💻Code: github.com/ABILiLab/OmniClea…
📜Paper: biorxiv.org/content/10.64898…
#ComputationalBiology #Bioinformatics #Proteomics #Protease #GraphNeuralNetworks #GeometricDeepLearning #StructuralBiology #AlphaFold #ProteinDesign #MachineLearning
9
70
3,782