
17/25 𝗙𝗹𝗲𝘅𝗶𝗯𝗹𝗲 𝗙𝗹𝗼𝘄𝘀 𝗳𝗼𝗿 𝗕𝗶𝗼𝗹𝗼𝗴𝗶𝗰𝗮𝗹 𝗦𝗲𝗾𝘂𝗲𝗻𝗰𝗲 𝗗𝗲𝘀𝗶𝗴𝗻
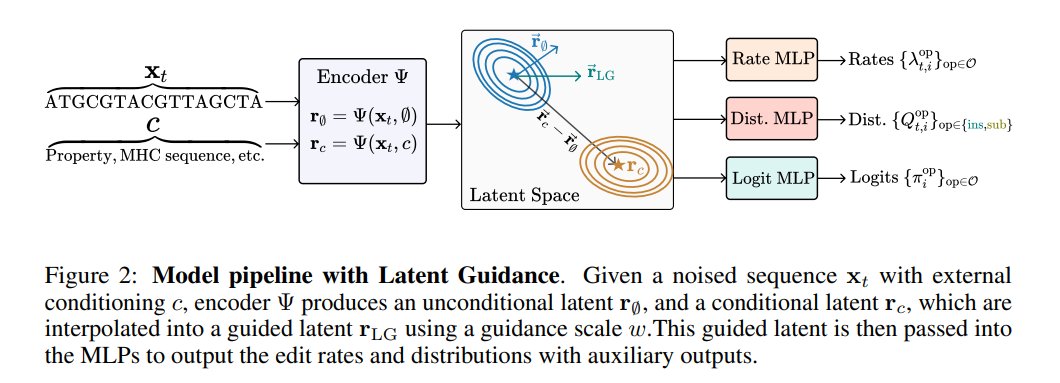
This paper addresses limitations in Discrete Flow Matching (DFM) for biological sequence design by proposing a structured coupling for domain-specific preferences and a latent edit-based rate parameterization for variable-length generation. It introduces a latent classifier-free guidance mechanism and Dirichlet-prior temperature scaling for test-time control, achieving state-of-the-art performance across diverse tasks including density estimation, unconditional/conditional DNA, and peptide sequence generation.
#DiscreteFlowMatching #BiologicalSequenceDesign #GenerativeModels #Bioinformatics #MachineLearning #DNAgeneration #PeptideGeneration
Paper Link: arxiv.org/abs/2606.10543

1
7
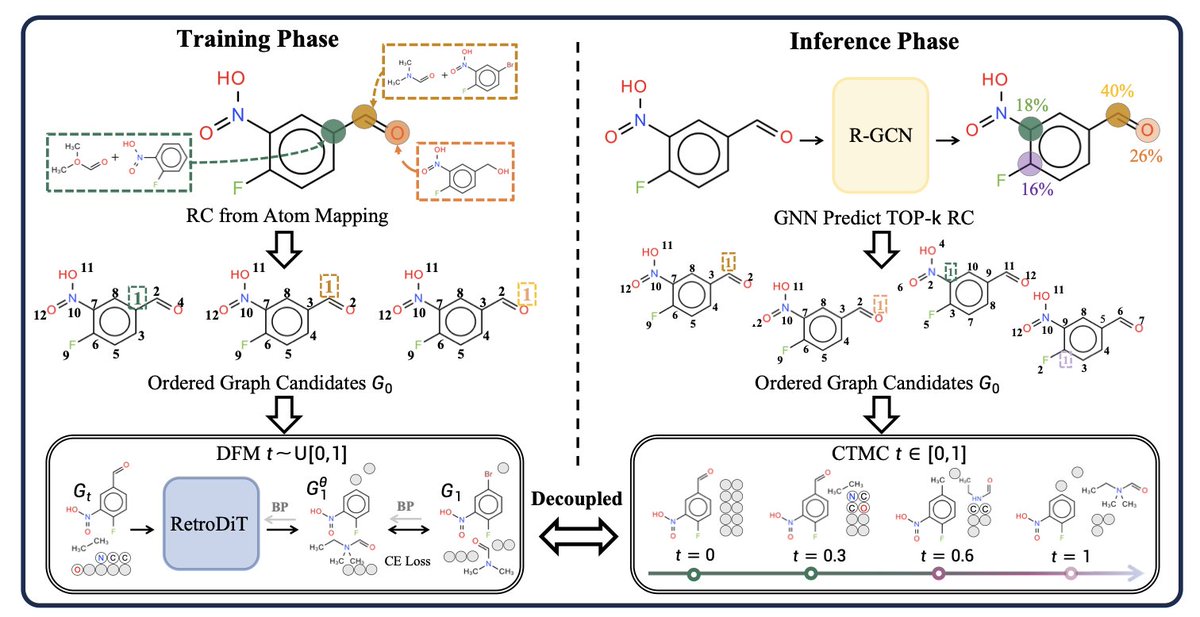
Order Matters in Retrosynthesis: Structure-aware Generation via Reaction-Center-Guided Discrete Flow Matching
1. A 280K-parameter model with proper atom ordering matches a 65M-parameter model without it—showing that structural inductive bias outperforms brute-force scaling in retrosynthesis prediction.
2. The key insight: atom ordering in neural representations matters for chemical reactions. By placing reaction center atoms at the sequence head, the method transforms implicit chemical knowledge into explicit positional patterns.
3. The proposed RetroDiT backbone combines rotary position embeddings (RoPE) with discrete flow matching, enabling generation in just 20-50 steps compared to 500 steps required by prior diffusion methods.
4. The framework achieves state-of-the-art performance on USPTO-50k (61.2% top-1 with predicted RCs, 71.1% with oracle RCs) and USPTO-Full (51.3% and 63.4% respectively), surpassing foundation models trained on 10 billion reactions while using orders of magnitude less data.
5. The modular design separates reaction center prediction from structure generation, allowing independent optimization and revealing that RC prediction is the primary performance bottleneck—providing clear direction for future improvements.
6. The method encodes the two-stage nature of retrosynthesis (identifying reaction centers then generating reactant structures) as a positional inductive bias, eliminating the need for explicit templates while maintaining generation flexibility.
7. With oracle reaction centers, the generative backbone reaches 71.1% on USPTO-50k, approaching RSGPT with 20× test-time augmentation (77.0%) without requiring massive pretraining or sampling tricks.
8. The reaction center definition covers eight transformation types including bond changes, charge changes, chirality changes, and hybridization changes—achieving over 99% coverage with just the top two types across both benchmark datasets.
📜Paper: arxiv.org/abs/2602.13136
#Retrosynthesis #Chemoinformatics #MachineLearning #AIforScience #GraphNeuralNetworks #DiscreteFlowMatching #MolecularGeneration #DrugDiscovery
4
23
1,537

13 Oct 2025
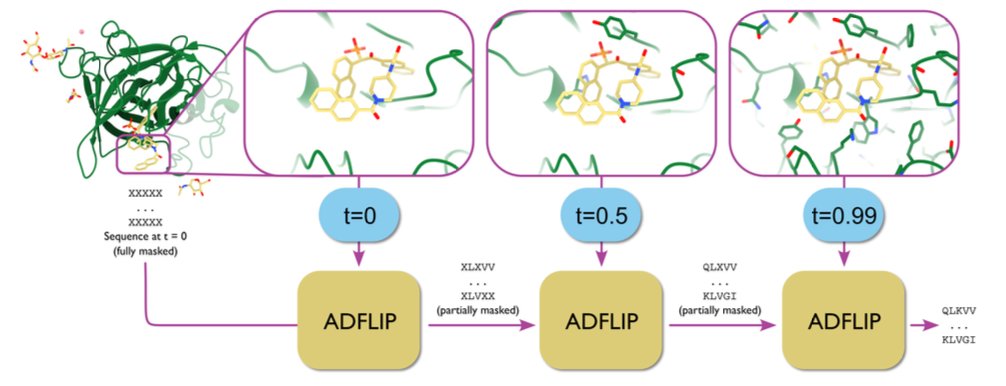
All-atom inverse protein folding through discrete flow matching
1. ADFLIP, a novel generative model for protein sequence design, has been introduced by Yi et al. This model leverages discrete flow matching to design protein sequences conditioned on all-atom structural contexts, including non-protein elements like ligands, nucleotides, and metal ions. It addresses the challenges of designing sequences for complex biomolecular assemblies and dynamic protein complexes.
2. ADFLIP incorporates predicted amino acid side chains progressively during sequence generation, providing crucial structural context that defines specific interactions with other molecules. This approach is particularly innovative for designing protein-ligand interactions and dynamic complexes with multiple structural states.
3. The model employs a multi-scale graph neural network as the denoising backbone, integrating both atom and residue-level information. This allows ADFLIP to capture detailed structural nuances, leading to state-of-the-art performance in single-structure and multi-structure inverse folding tasks.
4. ADFLIP implements training-free classifier guidance sampling, enabling the integration of arbitrary pre-trained models to optimize designed sequences for desired protein properties. This flexibility allows researchers to steer sequence generation towards specific outcomes without retraining the model.
5. The performance of ADFLIP was evaluated on protein complexes with small-molecule ligands, nucleotides, and metal ions, including dynamic complexes determined by NMR. The model demonstrated excellent potential for all-atom protein design, outperforming existing methods in sequence recovery and foldability.
6. ADFLIP’s ability to handle dynamic protein complexes through ensemble sampling across multiple structural states is a significant step forward in protein design. This capability is essential for designing proteins that undergo conformational changes during their functional cycles.
7. The training-free guidance sampling mechanism allows ADFLIP to leverage powerful existing regressors for guided generation, making it a versatile tool for protein design. This approach was demonstrated by guiding sequence generation towards higher predicted binding affinities using DSMBind.
📜Paper: raw.githubusercontent.com/ml…
#ProteinDesign #DiscreteFlowMatching #AllAtomModeling #Bioinformatics #MachineLearning
8
61
3,501