
Uncertainty Estimation for Molecular Diffusion Models
1. The paper addresses a practical gap in 3D molecular diffusion generation: pretrained diffusion models can output chemically invalid/unstable molecules, but they provide no principled per-sample signal of “this generation is likely low quality,” which is crucial when downstream evaluation (docking, wet lab) is expensive.
2. The authors propose a post-hoc uncertainty estimator that works with an existing pretrained molecular diffusion model (no retraining): fit a Laplace approximation around the denoiser’s MAP parameters and use it to quantify how variable the denoiser’s noise predictions are during sampling.
3. Core idea: for selected denoising timesteps, sample multiple parameter vectors from the approximate posterior q(θ), compute multiple noise predictions ε_t^m = f_{θ_m}(x_t, t), and take the elementwise sample variance across these predictions; then aggregate over timesteps, atoms, and feature dimensions into a single scalar uncertainty score per generated molecule.
4. The uncertainty is computed along the generation trajectory, motivated by the intuition that “internally uncertain” samples should induce more unstable/variable denoising behavior; empirically, only a small subset of timesteps is needed, reducing overhead.
5. On QM9, the resulting uncertainty score is informative of sample quality: it shows statistically significant negative Spearman correlations with molecular stability, atom stability, and validity, and it is consistently more predictive than diffusion negative log-likelihood (NLL) as a per-sample quality indicator.
6. Concrete QM9 correlations (Spearman ρ): for EDM, uncertainty vs. molecular stability is −0.284 (vs. NLL −0.150); for GeoLDM, −0.333 (vs. NLL −0.171). Similar gaps hold for atom stability and validity, suggesting likelihood is a weaker “verifier” than the proposed uncertainty for these quality metrics.
7. The paper then uses uncertainty for test-time scaling: oversample N molecules (10K→20K) and keep the 10K lowest-uncertainty samples. This improves stability/validity on QM9 for both EDM and GeoLDM, outperforming NLL-based filtering, with a modest tradeoff of ~1% drop in uniqueness.
8. The gains can be material relative to changing the base generator: for EDM on QM9, oversampling to 20K and filtering back to 10K yields ~10% molecular stability improvement, ~1% atom stability improvement, and ~5% validity improvement—comparable in magnitude to switching from EDM to GeoLDM at the same 10K budget.
9. Limitations and ablations: the filtering benefits do not transfer to GEOM-Drugs (larger, more complex molecules), where neither uncertainty- nor NLL-based filtering beats random subsampling. Ablations also show the Fisher-based Laplace covariance is not essential (isotropic perturbations around MAP perform similarly), implying the score may behave more like a sensitivity-to-perturbation measure than strict Bayesian epistemic uncertainty; signal concentrates near the clean end of the trajectory (late denoising steps).
📜Paper: arxiv.org/abs/2606.13451
#DiffusionModels #MolecularGeneration #ComputationalChemistry #UncertaintyEstimation #TestTimeScaling #BayesianDeepLearning #GenerativeModels #3DGeometry #QM9 #GEOMDrugs

1
15
1,131
DeepRHP: A Hybrid Variational Autoencoder for Designing Random Heteropolymers as Protein Mimics
1. The paper introduces DeepRHP, a semi-supervised hybrid VAE that learns latent representations of random heteropolymer (RHP) sequence ensembles while explicitly constraining the latent space to reflect function-related chemical features, aiming to make RHP design more data-driven than empirical screening.
2. Key architectural idea: a classical sequence VAE is paired with a parallel feature-based VAE that reconstructs a deterministic chemical feature y derived from the same sequence x; both branches share the same latent variable z, encouraging z to encode both sequence-pattern statistics and chemically meaningful structure.
3. The training objective modifies the standard VAE ELBO by combining two reconstruction terms: (a) discrete sequence reconstruction (cross-entropy over monomer tokens) and (b) feature reconstruction (MSE on y), weighted by a tunable α, while keeping the KL regularization on q(z|x) vs p(z).
4. The “feature” used for semi-supervision is the sliding-window average hydrophilic–lipophilic balance (HLB), motivated by prior evidence that local hydrophobicity/solubility patterning is strongly tied to RHP behavior in protein stabilization and transport applications.
5. Data pipeline: the study simulates 10,000 RHP sequences per monomer composition using Compositional Drift (copolymer models Monte Carlo), focusing on a 4-methacrylate monomer set (MMA, EHMA, OEGMA, SPMA) spanning hydrophobic, very hydrophobic, hydrophilic, and charged chemistries.
6. To connect synthetic polymers to biology, ~30k membrane and ~30k globular protein sequences (UniProt, 50% identity threshold) are reduced into a 4-letter “monomer-equivalent” alphabet based on residue hydrophobicity/charge, enabling joint embedding and similarity analysis between proteins and RHP ensembles.
7. Design insight 1 (alphabet size): by comparing 2-monomer vs 4-monomer RHP libraries in the learned latent space (visualized via PCA of latent factors), the paper argues that 2-monomer sequence space is too broad relative to protein-like regions, whereas 4-monomer libraries yield more localized, protein-overlapping distributions—supporting why four monomers can be “enough” for protein-mimic behavior.
8. Design insight 2 (composition): within a fixed 70% hydrophobic / 30% hydrophilic constraint, varying the MMA:EHMA ratio produces distinct RHP ensembles; DeepRHP’s latent-space overlap with Aquaporin Z (AqpZ) projections highlights specific compositions (notably matching the published optimal formulation) as most similar to the target membrane protein.
9. Practical takeaway: DeepRHP reframes RHP design as an ensemble-level representation learning problem—enabling composition suggestion by latent-space similarity to target proteins—without requiring exact polymer sequences, 3D structures, or multiple sequence alignment, and with a plug-in pathway to incorporate other chemical features beyond HLB.
10. The authors report ablations indicating the hybrid (feature-guided) architecture outperforms a classical VAE alone for producing useful latent structure, while noting that current evaluation is largely qualitative and motivating future quantitative metrics and downstream tasks (e.g., membrane protein subclass discrimination, RHP–protein similarity scoring).
📜Paper: arxiv.org/abs/2606.11651
#ComputationalBiology #MachineLearning #DeepLearning #VAE #GenerativeModels #PolymerScience #MaterialsInformatics #ProteinEngineering #MembraneProteins #Cheminformatics

2
14
1,027
Flexible Flows for Biological Sequence Design
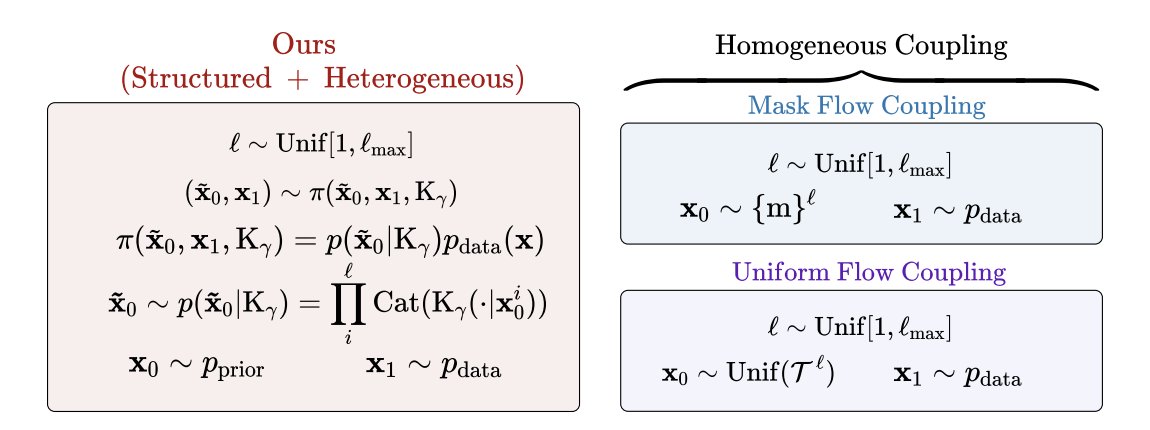
1. FlexFlow reframes discrete flow matching for biological sequences by changing the coupling (forward endpoint pairing) rather than the training objective: a structured, biology-informed coupling uses substitution matrices (e.g., BLOSUM for proteins; JC69/HKY85-style biases for nucleotides) to tilt the source distribution toward evolutionarily plausible neighborhoods.
2. The key idea is to keep the standard token-wise mixture path and CTMC machinery intact, but swap the usual “uninformative” couplings (uniform/masked) with a transition kernel Kγ that encodes preferred substitutions; when Kγ is uniform, the method reduces to the standard uniform coupling.
3. For variable-length generation, FlexFlow builds on Edit Flows by parameterizing reverse-time CTMC rates via edit operations (insertion, deletion, substitution). Instead of treating positions independently, it introduces a shared global latent r that conditions per-position edit decisions, coupling token-level operations through sequence-level context.
4. FlexFlow adds test-time control over edit behavior: operation probabilities are temperature-scaled and modeled with a Dirichlet prior over {ins, sub, del}. By changing Dirichlet concentrations α post-hoc, users can bias generation toward more insertions vs substitutions vs deletions without retraining, effectively acting as an “operation budget controller.”
5. The paper proposes latent classifier-free guidance (CFG) as an alternative to rate-space guidance: it performs CFG by interpolating conditional/unconditional latents (rc and r∅) in continuous space, then uses the guided latent to drive all edit operations jointly—aiming for more globally coherent conditioning than token-wise rate guidance.
6. The latent guidance has a probabilistic interpretation: under Gaussian conditional/unconditional latent encodings and sufficiency assumptions, the guidance direction corresponds to the score of an implicit classifier p(c|r), making the latent interpolation analogous to a gradient ascent step on log p(c|r).
7. Training uses an augmented alignment space with a blank token ε to make edit-based objectives tractable: alignments define edit sequences between endpoints, and a Bregman-divergence-style loss penalizes extraneous rates while rewarding edits that move xt toward x1.
8. DNA enhancer generation (unconditional, length 500) on fly brain and melanoma ATAC-seq datasets: FlexFlow achieves the best Fréchet Biological Distance among compared diffusion/flow baselines at the same sampling budget (100 reverse steps), and ablations indicate combining a frequency-informed prior with structured coupling performs best.
9. Conditional promoter design (human promoters, length 1024) conditioned on transcription initiation profiles: FlexFlow improves MSE of predicted regulatory activity versus prior baselines, with latent guidance outperforming rate guidance (reported 0.022 vs 0.024 MSE at 100 steps), suggesting benefits from global latent steering.
10. A new peptide–MHC II conditional generation benchmark is introduced using eluted ligand data with a strict split where no 9-mer is shared across train/test clusters. On this task, FlexFlow greatly improves a held-out DeepMHCII-based discriminator score (rate guidance 0.58; latent guidance 0.66), while highlighting a quality–diversity tradeoff (latent guidance can improve plausibility while worsening embedding-distance coverage metrics).
📜Paper: arxiv.org/abs/2606.10543
#ComputationalBiology #GenerativeModels #FlowMatching #DiffusionModels #ProteinDesign #DNADesign #PeptideDesign #MHC #MachineLearning #Bioinformatics
3
16
1,424

May 27
Scientific simulation has a brutal tradeoff:
fast is usually coarse,
accurate is usually expensive.
A new paper by Jiahe Huang, Sihan Xu, Sharvaree Vadgama, and Rose Yu proposes a serious way through that bottleneck:
Recursive Flow Matching.
The target is one of the hardest problems in scientific ML:
forecasting complex spatiotemporal dynamics without paying the full cost of traditional solvers or slow generative samplers.
Think fluid flow.
Climate fields.
Sea surface temperature.
Acoustic wave propagation.
High-dimensional systems where accuracy, stability, and speed all matter.
Diffusion-based emulators can be powerful, but they often require many sequential denoising steps.
Vanilla flow matching is faster, but aggressive step reduction can degrade fidelity, especially in long rollouts.
RecFM changes the geometry of the generation path.
Instead of learning one trajectory through time, it learns a family of recursively scaled trajectories that intersect at shared spatial states.
Then it enforces consistency across those scales.
That is the core move:
same state
different discretizations
aligned velocities
consistent trajectories
In plain English:
if a model reaches the same physical state through a large step or several smaller steps, its dynamics should agree.
This self-consistency reduces discretization error and stabilizes one- and few-step generation.
That matters because real-time scientific emulation cannot afford hundreds of sampling steps.
The paper reports that RecFM achieves high-fidelity 1-step and 2–4-step dynamic generation with performance comparable to state-of-the-art multi-step solvers.
Across scientific benchmarks, it reaches up to a 20× speedup over leading diffusion-based emulators while improving predictive accuracy, and reduces MSE by over 15% compared to vanilla flow matching.
On Helmholtz Staircase, the reported gains are especially striking: RecFM captures wave propagation patterns where the strongest baseline struggles, with much lower error.
The deeper lesson:
for scientific generative models, the future may not be “more steps.”
It may be better trajectory geometry.
RecFM suggests that if we enforce consistency across scales, we can preserve physical fidelity while collapsing inference into one or a few steps.
That is exactly the kind of capability needed for real-time emulation in climate, fluids, materials, and complex physical systems.
Full credit to the authors:
Jiahe Huang, Sihan Xu, Sharvaree Vadgama, Rose Yu.
Paper:
Recursive Flow Matching
arxiv.org/abs/2605.26535
Project:
jhhuangchloe.github.io/RecFM…
I’m attaching the first page because the abstract is worth reading closely.
The next generation of scientific emulators may not just simulate faster.
They may learn trajectories that remain coherent across time scales.
#AIResearch #GenerativeModels #PhysicsAI #MachineLearning #ClimateAI

1
5
23
1,154

Scientific simulation has a brutal tradeoff:
fast is usually coarse,
accurate is usually expensive.
A new paper by Jiahe Huang, Sihan Xu, Sharvaree Vadgama, and Rose Yu proposes a serious way through that bottleneck:
Recursive Flow Matching.
The target is one of the hardest problems in scientific ML:
forecasting complex spatiotemporal dynamics without paying the full cost of traditional solvers or slow generative samplers.
Think fluid flow.
Climate fields.
Sea surface temperature.
Acoustic wave propagation.
High-dimensional systems where accuracy, stability, and speed all matter.
Diffusion-based emulators can be powerful, but they often require many sequential denoising steps.
Vanilla flow matching is faster, but aggressive step reduction can degrade fidelity, especially in long rollouts.
RecFM changes the geometry of the generation path.
Instead of learning one trajectory through time, it learns a family of recursively scaled trajectories that intersect at shared spatial states.
Then it enforces consistency across those scales.
That is the core move:
same state
different discretizations
aligned velocities
consistent trajectories
In plain English:
if a model reaches the same physical state through a large step or several smaller steps, its dynamics should agree.
This self-consistency reduces discretization error and stabilizes one- and few-step generation.
That matters because real-time scientific emulation cannot afford hundreds of sampling steps.
The paper reports that RecFM achieves high-fidelity 1-step and 2–4-step dynamic generation with performance comparable to state-of-the-art multi-step solvers.
Across scientific benchmarks, it reaches up to a 20× speedup over leading diffusion-based emulators while improving predictive accuracy, and reduces MSE by over 15% compared to vanilla flow matching.
On Helmholtz Staircase, the reported gains are especially striking: RecFM captures wave propagation patterns where the strongest baseline struggles, with much lower error.
The deeper lesson:
for scientific generative models, the future may not be “more steps.”
It may be better trajectory geometry.
RecFM suggests that if we enforce consistency across scales, we can preserve physical fidelity while collapsing inference into one or a few steps.
That is exactly the kind of capability needed for real-time emulation in climate, fluids, materials, and complex physical systems.
Full credit to the authors:
Jiahe Huang, Sihan Xu, Sharvaree Vadgama, Rose Yu.
Paper:
Recursive Flow Matching
arxiv.org/abs/2605.26535
Project:
jhhuangchloe.github.io/RecFM…
I’m attaching the first page because the abstract is worth reading closely.
The next generation of scientific emulators may not just simulate faster.
They may learn trajectories that remain coherent across time scales.
#AIResearch #ScientificMachineLearning #GenerativeModels #PhysicsAI #MachineLearning #ClimateAI

1
6
18
1,159

May 26
🎓 Fully Funded PhD in Biostochastics (Sweden 🇸🇪)
💶 Fully funded 4-year PhD with competitive doctoral salary progression outstanding research environment and benefits
✅ Passionate about #ComputationalBiology #DeepLearning #BayesianStatistics 🧬🧠📊
✅ Highly recommend this advanced interdisciplinary #fullyfunded #PhDPosition within the Department of Microbiology, Tumor and Cell Biology at @karolinskainst 🇸🇪
📌 This #phdproject focuses on developing flexible yet interpretable statistical and deep learning models for complex biological systems
You’ll work on:
🔷 Developing new modeling frameworks that bridge mechanistic and data-driven approaches
🔷 Designing and implementing deep learning and generative modeling methods for biological systems
🔷 Working across ecology, phylogenetics, and protein generative modeling applications
🔷 Applying advanced Bayesian inference, stochastic processes, Gaussian Processes, and latent-variable models
🔷 Conducting computational experiments and collaborating with experimental biology labs
🔷 Publishing cutting-edge interdisciplinary research at the intersection of statistics, AI, and biology
🌍 Contribute to next-generation computational biology by developing interpretable and scalable models that advance understanding of complex living systems.
✅ Work with Dr. @BenjMurrell and the Biostochastics research environment at Karolinska Institutet
⏰ 𝗗𝗲𝗮𝗱𝗹𝗶𝗻𝗲: 𝟭𝟱𝘁𝗵 𝗝𝘂𝗻𝗲, 𝟮𝟬𝟮𝟲
👉 Full details & apply here:
🔗 phdscanner.com/opportunities…
📩 Want more like this?
➕ Follow @PhdScanner and join WhatsApp for updates:
whatsapp.com/channel/0029Vb5…
🌐 Visit: phdscanner.com
#fullyfundedPhD #PhDposition #KarolinskaInstitutet #Sweden #Biostochastics #ComputationalBiology #DeepLearning #GenerativeModels #BayesianStatistics #MachineLearning #Bioinformatics #ResearchOpportunity
@phdhardtalk
♻️ Share with someone applying this cycle

ALT https://www.phdscanner.com/opportunities/phd-vacancies-karolinska-institutet-sweden-doctoral-phd-student-position-in-biostochastics-78dffa25-3399-4ad9-94e9-11b4e62a55ef
1
10
546
Atom-level Protein Representation Learning Improves Protein Structure Prediction
1. The paper proposes TRIPROREP, a protein representation model that explicitly targets structure-predictive use cases (complex docking signals and structure-model supervision), rather than only downstream function annotation where sequence-only PLMs can remain competitive.
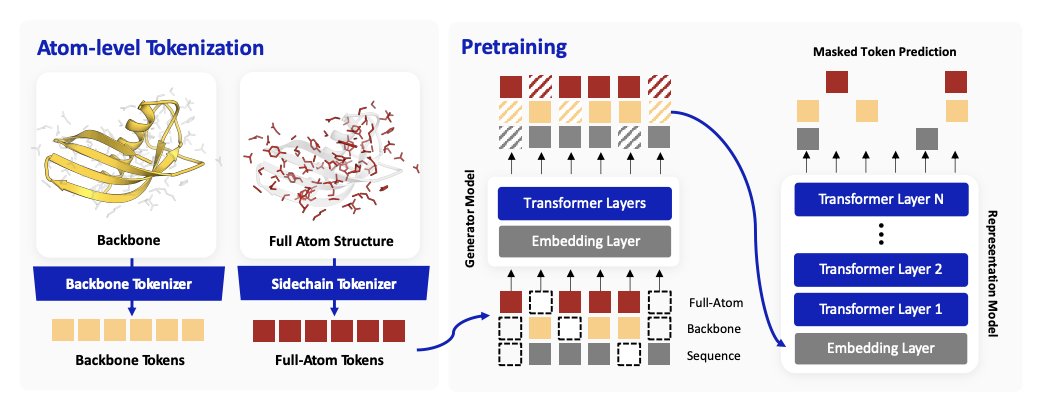
2. Core idea: learn a coupled residue-level representation from three aligned “views” of the same protein: amino-acid identity, backbone geometry tokens, and newly introduced full-atom residue geometry tokens that preserve side-chain/heavy-atom arrangements important for interfaces.
3. The full-atom tokenization is built with a residue-level VQ-VAE over Atom37 heavy-atom coordinates expressed in an SE(3)-invariant local frame (defined by N, Cα, C), plus dihedral/torsion features; a 512-entry codebook captures rotamer-sensitive local atomic states that backbone-only tokens largely miss.
4. Pretraining objective: ELECTRA-style generator corruption across all three token streams, followed by a discriminator trained to recover the original tokens (not just “replaced vs unchanged”). Independent corruption per view creates plausible-but-inconsistent cross-view combinations, forcing the model to learn cross-view consistency between sequence, backbone, and side-chain geometry.
5. Scale and data: TRIPROREP is pretrained at 35M, 150M, 650M, and 2.8B parameters on 83.7M predicted structures from AFDB (filtered by pLDDT) plus ESMAtlas, using 512-residue crops and a high masking probability per view to amplify corrective signals.
6. The paper also introduces REPSP, a structure-predictive benchmark built from 1.8M AFDB homodimer complexes (paired with corresponding apo monomers), split by 50% sequence-identity clusters, designed to test whether representations contain geometry useful for predicting complexes and supervising folding models.
7. REPSP Task 1 (homodimer co-folding / flexible docking): a modified SimpleFold-style flow-matching docking model takes frozen apo-chain representations and predicts holo homodimer structures. TRIPROREP improves interface and overall metrics across all sizes (e.g., 2.8B reaches DockQ 0.499 vs ESM2-3B 0.387; 650M already beats several “huge” baselines on most metrics).
8. REPSP Task 2 (per-residue interface property probing): simple MLP probes on frozen monomer representations predict homodimer-derived labels (binding site, ∆SASA, Levy tier, bond/interaction types). TRIPROREP consistently improves AUPRC/AUROC and correlation metrics, suggesting binding-relevant signals are encoded at residue resolution without fine-tuning.
9. REPSP Task 3 (representation-aligned distillation for monomer folding): pretrained representations are used as dense alignment targets (cosine similarity) to guide a structure predictor’s hidden states. TRIPROREP provides the strongest alignment target among large-scale models at 650M (e.g., TM-score 0.5822 vs baseline 0.5422), indicating its geometry is not only decodable but also useful as supervision.
10. Generalization and trade-offs: gains persist on real-world RCSB homodimers deposited after June 2023, and TRIPROREP remains competitive (not necessarily best) on conventional EC/GO function probing—supporting the paper’s thesis that structure-aware pretraining should be evaluated on structure-predictive tasks, not only function benchmarks.
💻Code: holymollyhao.github.io/TriPr…
📜Paper: arxiv.org/abs/2605.22133
#ProteinRepresentationLearning #ProteinStructure #StructuralBioinformatics #ComputationalBiology #DeepLearning #GenerativeModels #ProteinDocking #AlphaFold #ESMFold #VQVAE
9
53
2,727
LineageFlow: Flow Matching for High-Fidelity Family-Aware Protein Sequence Generation
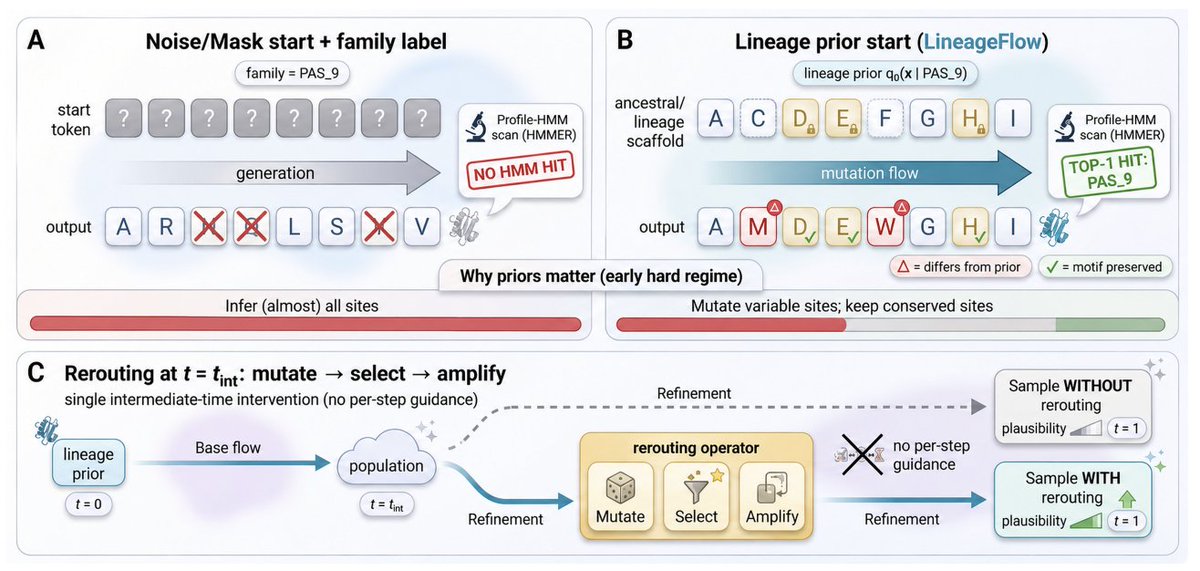
1. The paper argues that a key bottleneck in family-conditioned protein generation is the initialization prior: uniform-simplex noise or mask corruption erases evolutionary structure, forcing models to reconstruct conserved motifs “from scratch,” which weakens family control and plausibility.
2. LineageFlow replaces generic priors with lineage priors derived from ancestral sequence reconstruction (ASR): for each Pfam family, it infers a phylogeny from the MSA, performs marginal ASR at the root, and converts the site-wise root posterior into Dirichlet parameters used as a family-specific prior over the probability simplex.
3. With this design, generation is reframed as structured mutation from an evolved scaffold: conserved positions start concentrated, while variable sites retain uncertainty, aligning the trajectory with a family-specific manifold without feeding family labels or MSA prompts into the denoiser.
4. Methodologically, it builds on Dirichlet Flow Matching (DFM) on the simplex: each site follows an analytic Dirichlet path Dir(α(h,l) (tmax t) ei), with a derived lineage-specific vector field that conserves probability mass and keeps trajectories on the simplex.
5. Training uses a classifier parameterization: a transformer denoiser (initialized from ESM2) predicts terminal residues given (Xt, t), optimized by cross-entropy on valid (non-gap) MSA positions; the drift field is reconstructed by mixing analytic per-residue fields weighted by the predicted terminal distribution.
6. A second contribution is rerouting: a single intermediate-time inference intervention inspired by directed evolution (mutate → select → amplify) that steers samples toward a fitness objective without per-step gradient guidance, formalized as KL-regularized exponential tilting of the intermediate distribution.
7. Large-scale evaluation trains one shared model across 8,886 Pfam families (~8.94M sequences; 5% held-out per family) and scores generation by profile-HMM family validity (HMMER), foldability proxy (OmegaFold pLDDT), self-consistency (ESM-IF perplexity), novelty (MMseqs2 NN identity), and diversity (MMseqs2 clustering).
8. Results emphasize the role of priors: uniform-/mask-initialized baselines (DFM, EvoDiff) show essentially zero Pfam top-1 family accuracy under this strict HMM library scan, even when given explicit family labels; ASR prior alone (iid sampling) already yields high family validity, indicating ASR carries strong family signal.
9. LineageFlow with rerouting achieves near-natural family validity (Accfam 95.3% vs 96.6% for held-out natural sequences), improves foldability over prior-only and over several baselines (mean pLDDT 76.6), while keeping substantial novelty among foldable samples (Novelty@0.8 86.2%, Novelty@0.6 48.9%) and strong diversity.
10. A mechanistic analysis attributes gains to the “hard regime” at early times: Bayes-oracle denoising accuracy is higher under ASR priors than uniform priors when states are most corrupted, raising the recoverable signal ceiling and reducing early errors that propagate through the flow.
11. In a zero-shot enzyme case study, the denoiser is trained without three enzyme families, but priors are still built from their MSAs/trees; sampling without fine-tuning preserves motifs and novelty, and rerouting (using an unsupervised ESM2 plausibility objective) increases motif agreement and improves solubility/thermostability proxy distributions.
12. Limitations noted: reliance on high-quality MSAs and phylogenetic inference for priors; generation is tied to family alignment coordinates and does not model indels explicitly; evaluation relies on computational proxies (pLDDT, predictor-based properties) without experimental validation; rerouting adds compute and depends on the fitness function.
💻Code: github.com/Jinx-byebye/Linea…
📜Paper: arxiv.org/abs/2605.22252
#ComputationalBiology #ProteinDesign #GenerativeModels #FlowMatching #DiffusionModels #Phylogenetics #AncestralSequenceReconstruction #MachineLearning #Bioinformatics
7
32
2,856
AgForce Enables Antigen-conditioned Generative Antibody Design
1 Existing antigen-conditioned antibody design models often fail to actually use the antigen: across 11 CDR-H3 design methods on CHIMERA-BENCH, predictions at antigen-contacting positions recover worse than non-contacting positions, and an antigen-free baseline can achieve the strongest binding metrics—evidence of “antigen blindness.”
2 The paper identifies three causally linked failure modes behind this: antigen blindness (CDRs barely change across targets), vocabulary collapse (GNN greedy decoding predicts only ~3–5 amino acids per position vs native EV ~15.5), and a “cross-entropy ceiling” showing per-position cross-entropy optimizes to the positional marginal distribution, making antigen-specific predictions provably unattainable under the standard objective.
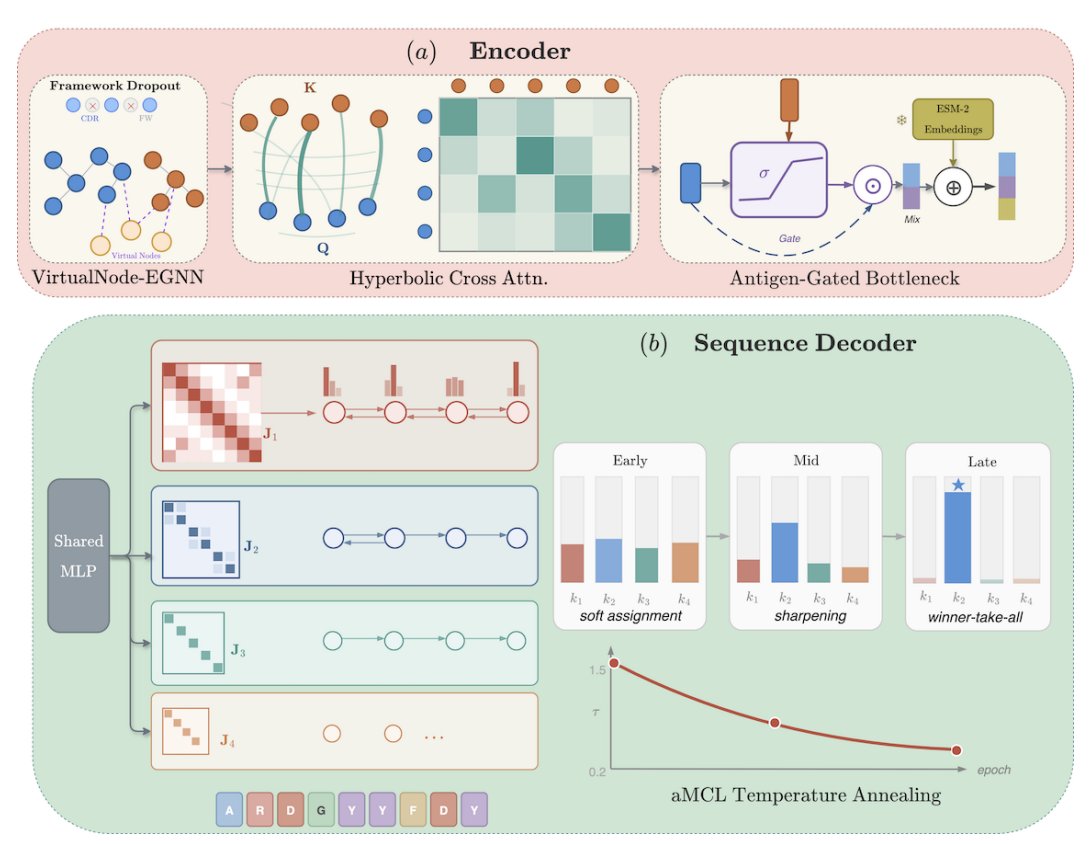
3 AGFORCE is proposed as an encoder-decoder co-design model that directly targets these failure modes rather than only changing the encoder: a VirtualNode-EGNN encoder (E(3)-equivariant) plus specialized conditioning and decoding mechanisms intended to block shortcut learning from the antibody framework.
4 To prevent the framework shortcut path (a key driver of antigen blindness), AGFORCE introduces framework dropout (randomly zeroing heavy-chain framework embeddings during training), plus gated bottlenecks and hyperbolic cross-attention between CDR and epitope embeddings to better transmit antigen information.
5 To break the cross-entropy ceiling and reduce vocabulary collapse, AGFORCE replaces the standard linear amino-acid head with an MDN-Potts sequence decoder: a mixture density network with K=4 components, augmented by Potts-like pairwise couplings between adjacent positions and decoded with belief propagation, enabling multi-modal sequence distributions beyond a single positional marginal.
6 Training uses annealed Multiple Choice Learning (aMCL): mixture components receive Boltzmann-weighted responsibility that anneals toward sharper assignments, encouraging component specialization; the paper argues this changes the optimal solution so components can deviate from the global positional marginal (i.e., can become antigen-conditional).
7 Diversity is explicitly regularized with a GDPP spectral loss to better match native distributional diversity (effective vocabulary and motif variety), addressing the observed collapse where rare but important residues (e.g., W/C/M) are nearly never predicted by greedy GNN baselines.
8 Antigen conditioning is further enforced with an antigen cycle-consistency style objective: an InfoNCE antigen classification loss computed from the predicted CDR probability distributions (a differentiable “soft sequence” embedding) against antigen embeddings, ensuring gradients flow through the sequence decoder and forcing predicted distributions to encode antigen identity.
9 On CHIMERA-BENCH (epitope-group split; 292 test complexes), AGFORCE reports improved sequence recovery and binding quality simultaneously: AAR 0.40 (vs 0.37 for strongest GNN baselines), lowest perplexity (2.95), and best interface metrics (fnat 0.67, iRMSD 1.30 Å, DockQ 0.74, epitope F1 0.77). It also increases antigen-specific diversity (95.5% unique sequences) and raises effective vocabulary to 9.4 (vs 3.0–5.5 for greedy GNN methods), though still below native diversity.
💻Code: github.com/mansoor181/ag-for…
📜Paper: arxiv.org/abs/2605.21610
#ComputationalBiology #ProteinDesign #AntibodyDesign #GenerativeModels #GraphNeuralNetworks #MachineLearning #Bioinformatics #StructuralBiology
5
22
1,910
Generating Physically Consistent Molecules with Energy-Based Models
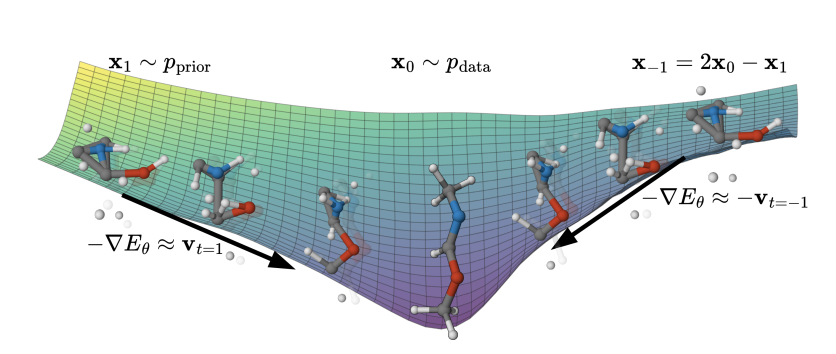
1. The paper introduces EBMol, a time-unconditional energy-based model for 3D molecule generation that explicitly learns a persistent scalar energy landscape Eθ(x), aiming to restore the Boltzmann-style inductive bias that diffusion/flow models typically avoid.
2. Core training idea: Restoring Field Matching (RFM), a simulation-free objective that shapes data configurations into local minima by training the negative energy gradient −∇Eθ to “point back” to data from both sides of an extended interpolation t ∈ [−1, 1] (interpolation from noise plus extrapolation beyond data), while enforcing stationarity at t = 0 via a smoothing schedule b(|t|) with b(0)=0.
3. Mixed state space modeling: molecules are represented as (c, p), where c are continuous 3D coordinates and p are simplex-constrained per-atom categorical distributions over elements; bonds are not modeled explicitly and are inferred post-hoc from geometry and types.
4. Architecture choice: an SE(3)-invariant energy is parameterized with an EGNN backbone and a permutation-invariant per-atom additive decomposition Eθ(c,p)=∑i Eθ(i)(c,p), enabling variable-sized molecules while keeping the energy physically interpretable as an atom-additive potential.
5. Practical stabilization in training: (i) optimal-transport alignment is used to match noise coordinates to data coordinates before linear interpolation, reducing path curvature; (ii) categorical interpolation uses a reflection rule to stay on the simplex; (iii) an energy anchoring regularizer pushes per-atom energies on clean data toward 0 to prevent arbitrary energy offsets across datapoints.
6. Sampling contribution: the work adapts the Mirror Langevin Algorithm (MLA) to jointly update coordinates (Euclidean Langevin) and atom types (mirror updates on the simplex using a negative-entropy mirror map, with softmax projection). This yields a single principled sampler for continuous discrete variables.
7. Inference-time compute scaling: parallel tempering is integrated with MLA, running multiple temperature chains with swap moves; more tempering rounds increase sample quality at the cost of more NFEs, producing a controllable quality–diversity tradeoff typical for energy-based sampling.
8. Benchmark results: EBMol reports state-of-the-art bond-implicit performance on QM9 and GEOM-Drugs, and shows strong improvements in “physical” metrics on GEOM-Drugs (bond lengths/angles/torsions vs GFN2-xTB-relaxed structures), including very low median relaxation energy at higher compute budgets.
9. Energy as a usable score: because Eθ is explicit, the learned energy can rank/filter samples; the paper shows EBMol energy correlates with relaxation energy decrease under GFN2-xTB for molecules generated by another model (EDM), suggesting Eθ is a practical first-pass physical quality metric without any energy supervision.
10. Composable and conditional generation without retraining: (i) shape-steered sampling is done by composing Eθ with differentiable shape potentials defined via PCA eigenvalues of atomic coordinates; (ii) zero-shot linker design is demonstrated by fixing fragments and sampling remaining atoms, achieving strong success rates across multiple tasks.
💻Code: github.com/griesbchr/EBMol
📜Paper: arxiv.org/abs/2605.18381
#EnergyBasedModels #MolecularGeneration #ComputationalChemistry #GenerativeModels #LangevinDynamics #GraphNeuralNetworks #QM9 #GEOMDrugs #MachineLearning
14
80
4,363

May 21
📝The Path from PCA to Autoencoders to Variational Autoencoders: Building Intuition for Deep Generative Modeling
👥by Alaa Tharwat & Mahmoud M. Eid
#PCA; #VariationalAutoencoder; #Autoencoder; #GenerativeModels
📖brnw.ch/21x2Hjz

1
1
27
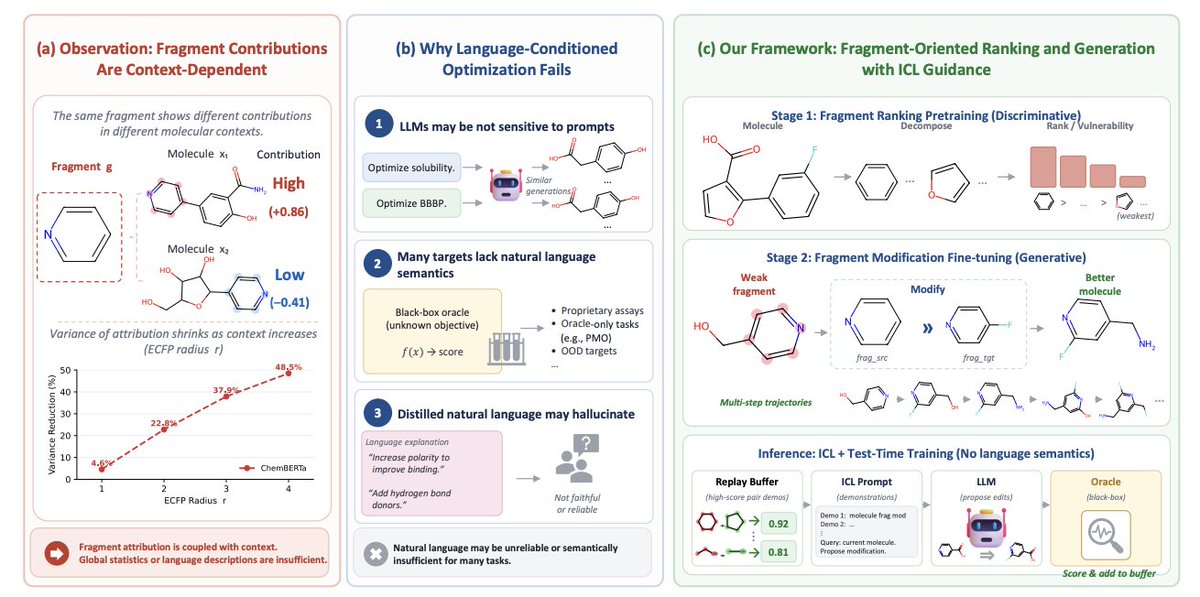
FORGE: Fragment-Oriented Ranking and Generation for Context-Aware Molecular Optimization
1. FORGE reframes molecular optimization as context-aware local editing: decide where to edit (fragment ranking) and how to edit (explicit fragment replacement), instead of rewriting full SMILES from a natural-language property prompt.
2. The paper motivates this shift with two findings: (i) prompt text weakly controls black-box optimization (swapping the true objective name to “QED” or “unknown property” changes PMO scores by <3%); (ii) fragment effects are strongly context-dependent, and grouping by local ECFP environments reduces attribution variance by ~8–11% vs shuffled controls.
3. FORGE is a two-stage supervised framework trained without expensive human text annotations or LLM-distilled chain-of-thought. It uses automatically mined, rule-verified low-to-high edit pairs to provide scalable, less hallucinatory supervision.
4. Stage 1 (discriminative) learns “where to edit” by ranking fragments under full-molecule context using fragment attribution labels. Labels come from (a) GNN mask-attribution for learned ADMET endpoints and (b) RDKit recomputation for rule-based properties (QED, logP, SA, TPSA, etc.), yielding ~2M training examples.
5. Stage 2 (generative) learns “how to edit” by generating explicit fragment replacements in the format: Modification: frag_src >> frag_tgt; Result: target_smiles (score). Supervision comes from two sources: SME context-conditioned edit pairs on 5 ADMET properties, plus ChEMBL matched molecular pairs oriented as low-to-high activity edits (with leakage filters for DRD2/JNK3/GSK3-β).
6. SME is introduced as a context-conditioned extension of SME: instead of aggregating fragment effects globally, it bins occurrences by attachment-atom ECFP radius-2 environments, searches for within-bin improving replacements, and falls back to global pools when bins are sparse—reducing label noise for local editing.
7. A key engineering detail is atom-level SMILES retokenization (QwenAtom) on top of Qwen3-0.6B: within <start smiles>…<end smiles>, a regex-based atomic parser enforces stable tokenization so the same fragment has consistent token identity across molecules (BPE would break this). This improves all 13 held-out Stage-1 subtasks, especially ICL ranking/vulnerability.
8. Inference targets black-box objectives via a replay-buffer in-context learning loop: the model conditions on demonstrations sampled from past (molecule, edit, new molecule, oracle score) tuples, proposes new fragment edits, queries the oracle, and updates the buffer—deriving task semantics from feedback rather than prompt semantics.
9. Results highlight the advantage under strict similarity constraints: on Prompt-MolOpt, FORGE degrades much more gracefully as similarity thresholds tighten (SUM 4.59/4.30/2.88 at δ=0/0.4/0.6), far above the prompt-based baseline at high δ. An ablation shows context-conditioned SME data is a major driver of gains when edits must stay local.
10. Despite using only a 0.6B backbone, FORGE outperforms or matches larger models and graph baselines across benchmarks: PMO-1k (1k oracle calls) reaches SUM 12.42 over 22 tasks; explicit modification output beats end-to-end SMILES rewriting on QED-DRD2; on ChemCoTBench it exceeds Qwen-3-8B (SFT) and GPT-4o on success rates, especially on real-target tasks (DRD2/JNK3/GSK3-β).
📜Paper: arxiv.org/abs/2605.10230
#ComputationalBiology #Chemoinformatics #MoleculeOptimization #DrugDiscovery #LLM #MachineLearning #GenerativeModels #ADMET #BlackBoxOptimization #SMILES
2
14
1,227
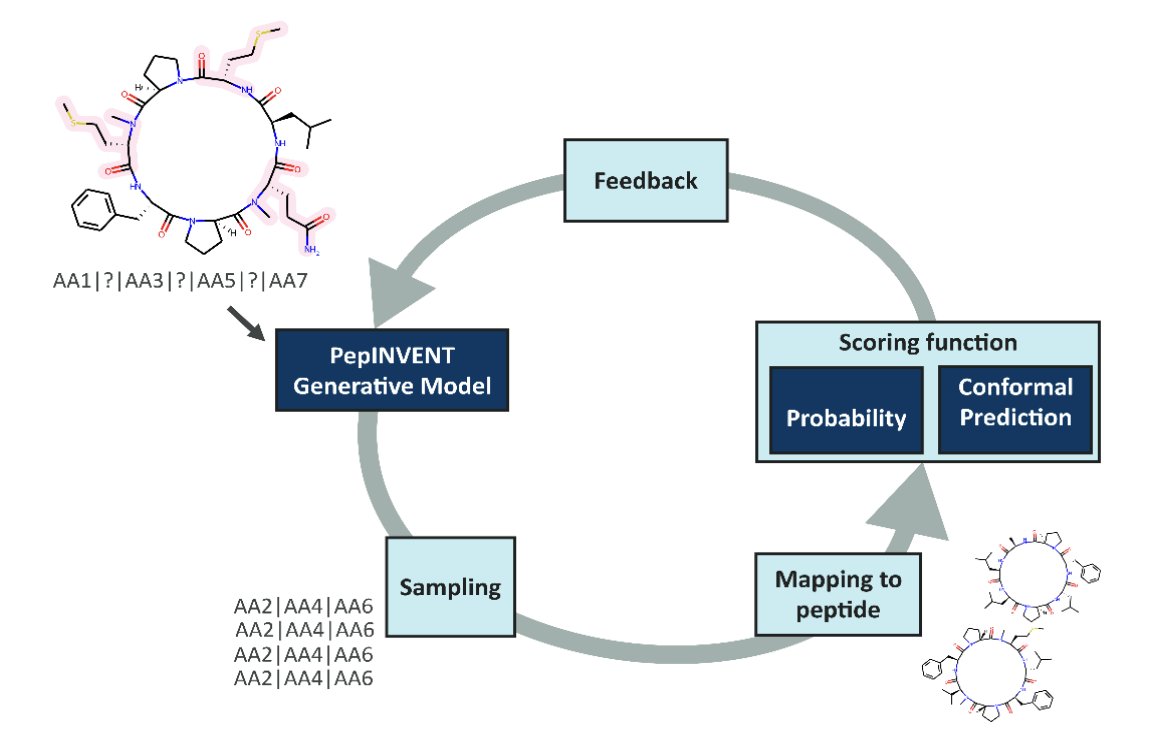
Confidence Is the Key: How Conformal Prediction Enhances the Generative Design of Permeable Peptides
1. The paper argues that RL-guided generative design can be misled by predictive models when generation drifts outside the model’s applicability domain, producing “high reward, high uncertainty” peptides—especially problematic for understudied cyclic peptides.
2. They integrate conformal prediction (CP) directly into the reinforcement learning (RL) scoring loop (PepINVENT-style peptide completion), so the agent is rewarded not just for predicted permeability, but for permeability predictions made with calibrated confidence at a user-chosen confidence level (here, 80%).
3. Core technical setup: a permeability classifier (XGBoost on ECFP features) is trained on CycPeptMPDB PAMPA data (6876 cyclic peptides; threshold LogPexp ≥ -6 as permeable). On top of this, they build an aggregated Mondrian inductive conformal predictor (ACP with 10 ICPs) outputting two p-values: P1 (permeable) and P0 (non-permeable).
4. Key conceptual point: CP’s two p-values encode “evidence for each class,” enabling four outcomes (Class 0, Class 1, Both, None). In RL, the target is not merely “high P(permeable)” but conformal efficiency: confidently permeable designs where P1 > 0.2 and P0 < 0.2 (at significance 0.2).
5. Baseline finding: optimizing raw model probability (standard practice) increases average predicted permeability (raw score rises ~0.51→0.87 over 350 epochs), but many “permeable” designs are not conformally confident—highlighting a mismatch between probability-based rewards and calibrated reliability.
6. They test multiple CP-based reward designs: maximize P1, maximize (1−P0), maximize (P1−P0), plus two discrete schemes: “harsh” (reward 1 only if both thresholds met) and “soft” (reward 1 if both met, 0.5 if one met, else 0).
7. Main methodological takeaway: single p-value optimization (P1 alone or 1−P0 alone) is learnable but does not reliably increase the number of confidently permeable peptides, because maximizing P1 does not ensure low P0 (and vice versa). The joint decision structure of Mondrian ICP matters.
8. Best-performing strategy: the CP “soft” scoring function converges fastest to the desired region (defined as reaching ~50% conformally efficient permeable predictions among valid molecules) and yields more reliable hits than raw-probability scoring when “hits” are defined as confident within-domain.
9. Practical insight on generation dynamics: the soft reward reduces brittleness from sparse rewards (compared to harsh) and improves efficiency—fewer unique valid molecules may be generated overall, but a higher fraction meet the calibrated confidence criterion, meaning less wasted exploration in uncertain space.
10. Robustness check: performance depends on peptide length and training-data coverage. The CP-soft approach works well for lengths well represented in training (6, 7, 8, 10), but deteriorates for 9, 11, 12, effectively flagging when the predictor’s applicability domain is being exceeded—useful as a “stop relying on this objective” signal.
📜Paper: arxiv.org/abs/2605.05770
#ConformalPrediction #ReinforcementLearning #GenerativeModels #PeptideDesign #CyclicPeptides #UncertaintyQuantification #Cheminformatics #ComputationalBiology #DrugDiscovery #MachineLearning
2
5
15
1,876
Expanding Functional Protein Sequence Space Using High Entropy Generative Models
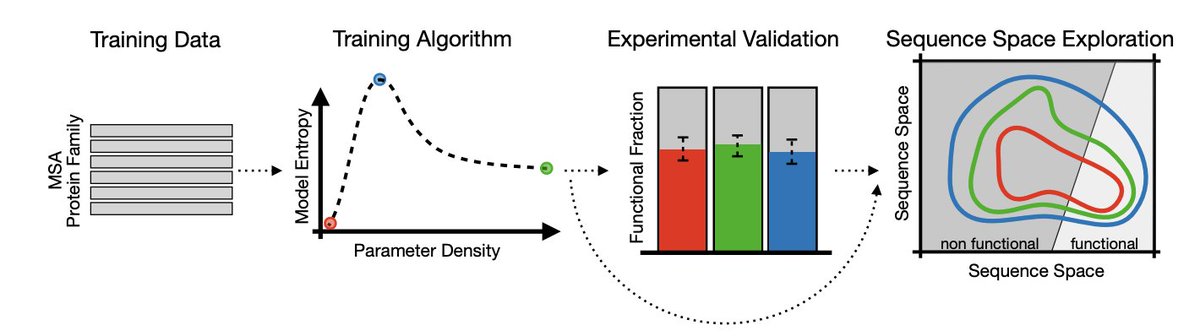
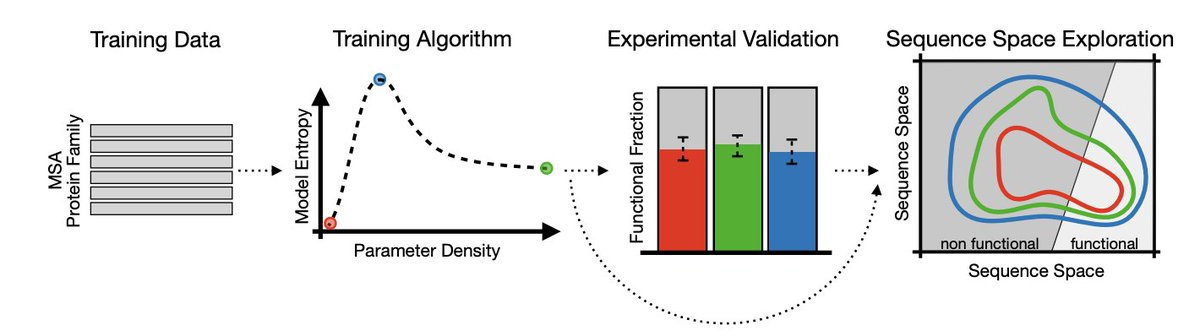
1. The study shows that “how wide” a protein generative model’s functional landscape is can differ massively even when different models generate similar fractions of functional enzymes in experiments; Shannon entropy is used as the key quantitative lens for this distinction.
2. Using the AroQ chorismate mutase family, the authors compare four Potts/Boltzmann-machine DCA variants: a fully connected model (bmDCA), a sparse model built by edge activation (eaDCA), a sparse model built by edge decimation (edDCA), and a maximum-entropy model (meDCA) chosen at the entropy peak along the decimation path.
3. Core technical point: along progressive pruning (decimation), entropy is non-monotonic—initial pruning removes noisy/weak constraints and increases entropy (better generalization), but over-pruning forces the remaining couplings to become overly rigid to maintain fit, causing an abrupt entropy collapse (overfitting via sparsity).
4. The selected meDCA model reaches high entropy at moderate parameter density (~12.5%) while retaining strong agreement with empirical pairwise connected correlations (Pearson ~0.94), highlighting that “fit quality” alone does not determine the breadth of the learned distribution.
5. Experimental validation (E. coli in vivo complementation) shows that all architectures—dense and sparse—can generate functional chorismate mutases, even at substantial divergence from natural sequences (tested bins: 20–25%, 40–45%, 60–65% divergence from closest natural homolog).
6. Despite broadly comparable functional hit rates across models, entropy differences are dramatic: the entropy gap between the highest-entropy and lowest-entropy models implies an effective viable sequence space ratio of about e^(ΔS) ~ 10^16, meaning some models explore a functional-capable region that is many orders of magnitude larger.
7. Cross-model scoring reveals an inclusion asymmetry: low-entropy models assign low probability to many sequences generated by meDCA, while meDCA assigns high probability to sequences generated by low-entropy models—consistent with meDCA covering a broader viable region that contains the narrower low-entropy regions as subsets.
8. The paper argues high-entropy models reduce overfitting and better represent local “neutral spaces” around natural proteins: leave-one-group-out tests show meDCA assigns systematically higher probability to withheld, distant natural sequences than bmDCA, indicating better generalization beyond the training neighborhood.
9. Local landscape geometry is probed via context-dependent entropy (CDE) as a mutability proxy: meDCA assigns higher mutability to natural sequences than low-entropy sparse models, aligning with the biological expectation that functional proteins sit in sizable neutral networks rather than isolated peaks; meDCA also mixes faster in MCMC sampling, suggesting smoother barriers between high-probability regions.
10. Broader implication: if the goal is merely to obtain functional sequences, multiple DCA architectures may suffice; but if the goal is to map or exploit the evolutionary fitness landscape broadly (diversity, neutral-space coverage, reduced overfitting), selecting for high entropy (as in meDCA) becomes a principled model-selection criterion, and the same entropy-vs-fit tension may matter for other architectures (the authors note similar entropy-collapse behavior with Transformers under certain stopping criteria).
💻Code: github.com/robertonetti/entr… ; github.com/spqb/adabmDCA ; github.com/robertonetti/high…
📜Paper: arxiv.org/abs/2605.03578
#ComputationalBiology #ProteinDesign #GenerativeModels #DCA #BoltzmannMachine #MaximumEntropy #Coevolution #FitnessLandscape #SyntheticBiology #MachineLearning
15
4,243
Expanding Functional Protein Sequence Space Using High Entropy Generative Models
1. The study shows that “how wide” a protein generative model’s functional landscape is can differ massively even when different models generate similar fractions of functional enzymes in experiments; Shannon entropy is used as the key quantitative lens for this distinction.
2. Using the AroQ chorismate mutase family, the authors compare four Potts/Boltzmann-machine DCA variants: a fully connected model (bmDCA), a sparse model built by edge activation (eaDCA), a sparse model built by edge decimation (edDCA), and a maximum-entropy model (meDCA) chosen at the entropy peak along the decimation path.
3. Core technical point: along progressive pruning (decimation), entropy is non-monotonic—initial pruning removes noisy/weak constraints and increases entropy (better generalization), but over-pruning forces the remaining couplings to become overly rigid to maintain fit, causing an abrupt entropy collapse (overfitting via sparsity).
4. The selected meDCA model reaches high entropy at moderate parameter density (~12.5%) while retaining strong agreement with empirical pairwise connected correlations (Pearson ~0.94), highlighting that “fit quality” alone does not determine the breadth of the learned distribution.
5. Experimental validation (E. coli in vivo complementation) shows that all architectures—dense and sparse—can generate functional chorismate mutases, even at substantial divergence from natural sequences (tested bins: 20–25%, 40–45%, 60–65% divergence from closest natural homolog).
6. Despite broadly comparable functional hit rates across models, entropy differences are dramatic: the entropy gap between the highest-entropy and lowest-entropy models implies an effective viable sequence space ratio of about e^(ΔS) ~ 10^16, meaning some models explore a functional-capable region that is many orders of magnitude larger.
7. Cross-model scoring reveals an inclusion asymmetry: low-entropy models assign low probability to many sequences generated by meDCA, while meDCA assigns high probability to sequences generated by low-entropy models—consistent with meDCA covering a broader viable region that contains the narrower low-entropy regions as subsets.
8. The paper argues high-entropy models reduce overfitting and better represent local “neutral spaces” around natural proteins: leave-one-group-out tests show meDCA assigns systematically higher probability to withheld, distant natural sequences than bmDCA, indicating better generalization beyond the training neighborhood.
9. Local landscape geometry is probed via context-dependent entropy (CDE) as a mutability proxy: meDCA assigns higher mutability to natural sequences than low-entropy sparse models, aligning with the biological expectation that functional proteins sit in sizable neutral networks rather than isolated peaks; meDCA also mixes faster in MCMC sampling, suggesting smoother barriers between high-probability regions.
10. Broader implication: if the goal is merely to obtain functional sequences, multiple DCA architectures may suffice; but if the goal is to map or exploit the evolutionary fitness landscape broadly (diversity, neutral-space coverage, reduced overfitting), selecting for high entropy (as in meDCA) becomes a principled model-selection criterion, and the same entropy-vs-fit tension may matter for other architectures (the authors note similar entropy-collapse behavior with Transformers under certain stopping criteria).
💻Code: github.com/robertonetti/entr… ; github.com/spqb/adabmDCA ; github.com/robertonetti/high…
📜Paper: arxiv.org/abs/2605.03578
#ComputationalBiology #ProteinDesign #GenerativeModels #DCA #BoltzmannMachine #MaximumEntropy #Coevolution #FitnessLandscape #SyntheticBiology #MachineLearning
2
1,071
Generative structure search for efficient and diverse discovery of molecular and crystal structures
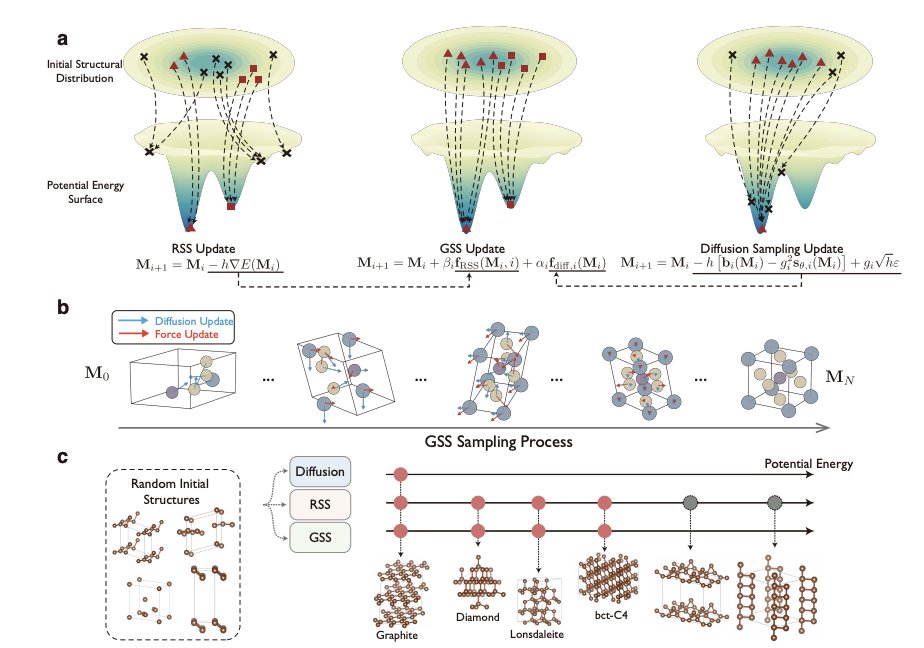
1 GSS (Generative Structure Search) unifies diffusion-based generation and random structure search (RSS) as two limits of the same iterative update process, then interpolates between them to get both: diffusion’s fast access to low-energy regions and RSS’s physically grounded convergence to local minima.
2 Core update: Mi 1 = Mi αi fdiff,i(Mi) βi fRSS(Mi), where fdiff comes from a pretrained diffusion score model and fRSS is driven by the potential energy surface (PES) gradient (forces). αi decays while βi increases (sigmoid schedule), so early steps use learned priors for global guidance and late steps enforce energy-based relaxation for physical validity.
3 Conceptual payoff: diffusion models can be biased toward training modes (often near a single global minimum) and miss rare-but-real metastable minima; RSS can cover many basins but wastes many trials in high-energy regions. GSS is designed to reduce wasted sampling while still recovering diverse metastable structures.
4 Periodic crystals: the method operates on fractional coordinates X (on a hypertorus due to periodicity) and lattice L. Guidance requires converting ML force-field outputs (Cartesian forces and virial stress) into gradients w.r.t. X and L, enabling energy-based steering during diffusion sampling in a consistent periodic parameterization.
5 Evaluation protocol: 12 representative periodic systems (elemental → quaternary, including an out-of-training-distribution ternary AlPN). They generate 1,024 samples per method and compare against an “exhaustive RSS” reference set of stable structures. Metrics: (i) coverage of reference minima (recall-like), (ii) efficiency via low-energy fraction / average energy (precision-like), plus a “budget cost” to solve a system (find all reference structures).
6 Main results on periodic systems: GSS consistently improves coverage over vanilla diffusion and improves energy efficiency over RSS. In coverage–efficiency space, GSS advances the Pareto front relative to the two baselines, rather than merely mixing their outputs.
7 Practical cost: when requiring recovery of all reference stable structures, GSS reduces the sampling budget by more than an order of magnitude compared with RSS, while vanilla diffusion saturates below full coverage on several systems.
8 Periodic-table scale test: elemental crystals with atomic number < 89 (excluding noble gases and trivial gaseous elements). GSS improves both low-energy yield and coverage broadly, with notable gains in groups where multiple competing structures are common (e.g., alkaline earth metals and carbon-group elements).
9 Case study (carbon polymorphs): diffusion tends to concentrate on graphite and can miss diamond/lonsdaleite/bct-C4; GSS recovers the diverse polymorph set while largely avoiding the high-energy invalid structures that RSS can produce.
10 Molecular conformers (aspirin): a diffusion model trained on one conformer mode collapses to that mode; RSS from random atoms fails to assemble the correct isomer/topology. GSS uses diffusion as an “oracle” for the correct molecular graph/topology manifold, then uses PES guidance to explore multiple stable conformers (recovering four minima in their torsion-angle landscape).
💻Code: github.com/Yifang-Qin/Genera…
📜Paper: arxiv.org/abs/2604.27636
#ComputationalChemistry #MaterialsScience #CrystalStructurePrediction #DiffusionModels #GenerativeModels #MolecularModeling #MLForceFields #StructureSearch #AI4Science
4
36
2,605
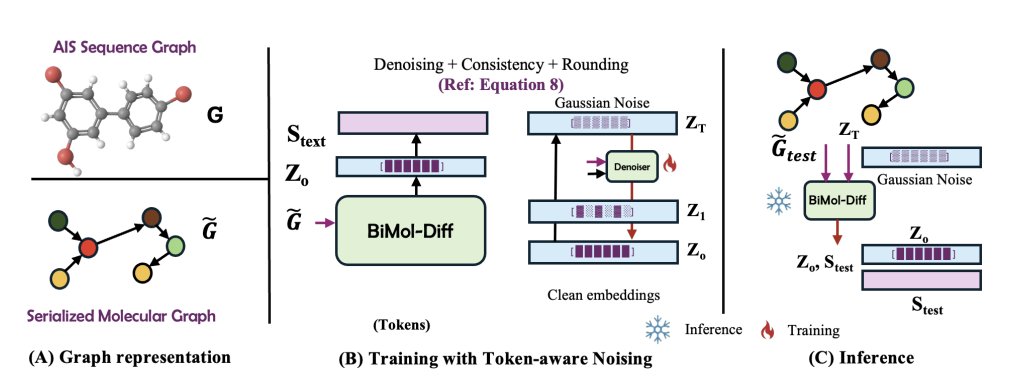
BiMol-Diff: A Unified Diffusion Framework for Molecular Generation and Captioning
1. BiMol-Diff proposes a single diffusion-based framework that handles both text→molecule generation and molecule→text captioning, aiming to make iterative molecule design loops more consistent than task-specific, separately trained systems.
2. The central technical idea is a token-aware noise schedule: instead of corrupting every position uniformly (as standard diffusion does), the model assigns position-dependent corruption based on how hard each token is to recover, using per-token denoising loss as a proxy for “difficulty”.
3. Mechanistically, the method periodically estimates token-wise difficulty profiles across diffusion timesteps, maps these difficulties to token-specific cumulative noise schedules via a piecewise-linear function, and enforces a valid monotone schedule using non-increasing isotonic projection.
4. Molecular representation is designed to reduce brittleness to SMILES syntax: molecules are tokenized with Atoms-in-SMILES (AIS) and also converted into a serialized “knowledge-graph-like” edge list of atom–bond–atom triplets using special tokens ([HEAD], [REL], [TAIL], [SEP]). Text↔molecule is modeled as text↔serialized-graph.
5. Training uses a z0-prediction diffusion objective (predict the clean latent at every step) plus two additional terms: a consistency loss for the first denoising step and a trainable rounding term that converts continuous latents back into discrete tokens. Inference further uses a “clamping” trick (nearest-neighbor projection onto the embedding table) to reduce rounding drift during intermediate steps.
6. On molecule captioning (M3-20M subset, ~360k pairs), BiMol-Diff reports best BLEU and BERTScore among compared baselines, with BLEU 0.567, ChrF 0.734, BERTScore-F1 0.843, MAUVE 0.925, while using fewer parameters than several AR and diffusion baselines in the table.
7. On text→molecule generation (ChEBI-20, 33,010 molecules), BiMol-Diff improves Exact Match to 0.262 vs 0.227 for the strongest compared AR adapter and diffusion baseline ( 15.4% relative), and improves fingerprint similarities (MACCS/RDKit/Morgan) while keeping high validity (0.901).
8. Ablations attribute a large portion of captioning gains to token-aware noising: switching from a uniform sqrt schedule to token-aware linear mapping improves BLEU (0.495→0.567), ChrF (0.682→0.734), and METEOR (0.531→0.626). AIS tokenization also outperforms regex and atom-level tokenization under the same schedule.
9. Efficiency analysis highlights a practical trade-off typical for diffusion: quality is strongest around 1000–2000 reverse steps; aggressive step reduction sharply degrades BLEU. Compared with DiffuSeq at 2000 steps, BiMol-Diff is faster per batch (89s vs 317s on V100) while also improving quality.
📜Paper: arxiv.org/abs/2604.24089
#moleculargeneration #diffusionmodels #cheminformatics #computationalbiology #nlp #multimodal #drugdiscovery #generativemodels
2
20
1,469

Apr 21
🚀 Excited to share our ICLR2026 @iclr_conf work on AI for molecular dynamics generation — a collaboration between Stanford University @Stanford and Lambda @LambdaAPI !
📄 "Align Your Structures: Generating Trajectories with Structure Pretraining for Molecular Dynamics"
🔗 Paper: arxiv.org/pdf/2604.03911v1
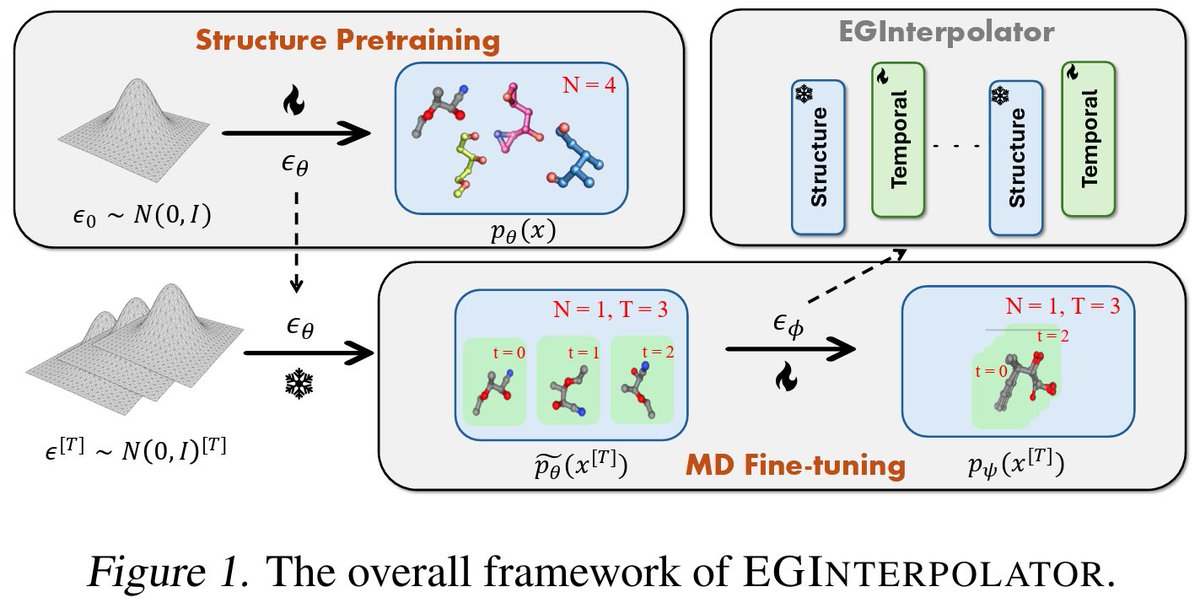
🧬 Learning molecular dynamics (MD) trajectories is challenging due to the scarcity and high dimensionality of trajectory data.
💡This work proposes EGInterpolator, a framework that leverages structure pretraining to overcome this bottleneck:
•🧬Pretrains on large-scale molecular structure datasets
•⏳Introduces an equivariant temporal interpolator to model trajectory evolution
•🔄Blends structure-based predictions with temporal modeling via learnable mixing
•📈Enables more stable and data-efficient trajectory generation
Key results:
🚀 Improved trajectory generation quality across QM9 and GEOM-Drugs
🧠 Better modeling of geometric, dynamical, and energetic distributions
⚡ Strong performance gains in forward simulation and interpolation tasks
🔁 Generalizes to peptides and preliminary protein settings
Takeaway:
✨Leveraging abundant molecular structures can significantly reduce the difficulty of learning molecular dynamics trajectories.
🙌 Huge shoutout to my Stanford collaborators: @aniketh_iyengar, @jiaqihan99, Pengwei Sun, @MingjianJ , and @StefanoErmon
#AI4Science #MolecularDynamics #MD #GenerativeModels #ICLR2026 #neurips #icml #DrugDesign #GeometricDiffusion #StructurePretraining
2
250
An integrated deep-learning framework for peptide-protein interaction prediction and target-conditioned peptide generation with ConGA-PepPI and TC-PepGen
1. The preprint presents an end-to-end, screening-oriented pipeline that links (i) partner-aware peptide–protein interaction prediction plus residue-level binding-site localization (ConGA-PepPI) with (ii) target-conditioned peptide sequence generation (TC-PepGen), aiming to connect prioritization, interpretability, and candidate expansion in one framework.
2. ConGA-PepPI is built around three practical PepPI constraints: peptides vs proteins are asymmetric partners, binding is conditional on the paired target context, and residue-level interface labels are scarce. The model addresses these with asymmetric dual-branch feature extraction, bidirectional cross-attention between peptide and protein, and progressive transfer from pair-level prediction to residue localization.
3. A key design choice is “partner-aware” representation learning: instead of encoding peptide and protein independently and fusing late, ConGA-PepPI uses serial/bidirectional cross-attention so peptide features update protein features and vice versa, encouraging conditional pair representations that better reflect PepPI recognition.
4. Another core component is progressive transfer learning: the model is first trained on larger sequence-level interaction labels, then fine-tuned for binding-site identification using fewer residue-annotated complexes, leveraging abundant pair supervision to improve interpretability.
5. ConGA-PepPI also adds supervised contrastive learning during sequence-level training to increase separability of interacting vs non-interacting pairs in latent space; ablations indicate this regularization provides the strongest final gain, particularly improving precision and MCC.
6. In five-fold cross-validation, ConGA-PepPI reports ACC 0.839 and AUROC 0.921 for binary interaction prediction (AUPR 0.915). Ablations show stepwise improvements from adding cross-attention, asymmetric branches, gated fusion, and contrastive learning (ACC 0.808 → 0.839; AUROC 0.885 → 0.921).
7. For binding-site localization, ConGA-PepPI reports protein-side AUPR 0.601 (challenging due to long sequences and class imbalance) and peptide-side AUPR 0.950 with AUROC 0.922, suggesting the transferred pair representation remains informative for residue-level interpretation—especially on the peptide side.
8. TC-PepGen targets a common failure mode in conditional generation: conditioning injected only once can dilute during autoregressive decoding. It preserves target information by injecting target-protein representations via cross-attention throughout decoding (layerwise conditioning), using a frozen protein encoder with an autoregressive decoder.
9. Under a controlled length-conditioned benchmark (native peptide length given), TC-PepGen peptides exceed native templates (hit defined as generated ipTM > native ipTM) in 40.39% of cases using AlphaFold 3 ipTM (also outperforming PepMLM and RFdiffusion under Chai-1 and ESMFold-based evaluation). In unconstrained generation (no true length), 27.09% still exceed native templates by AlphaFold 3, with generated lengths concentrated near the reference (often 0 to 5 residues).
10. The paper demonstrates integration in design workflows: generate many candidates for a target (e.g., GSK-3 beta), rank with ConGA-PepPI, then structurally evaluate with AlphaFold 3; top-ranked candidates show better structure-based scores than bottom-ranked ones. Additional case studies include in silico alanine scanning for hotspot identification and a low-homology MDM2 example where a novel peptide achieves AlphaFold 3 ipTM comparable to a known binder despite near-zero sequence identity.
📜Paper: arxiv.org/abs/2604.18467
#ComputationalBiology #PeptideDesign #ProteinEngineering #DeepLearning #ProteinLanguageModels #PeptideProteinInteractions #GenerativeModels #AlphaFold3 #DrugDiscovery

1
4
11
1,428
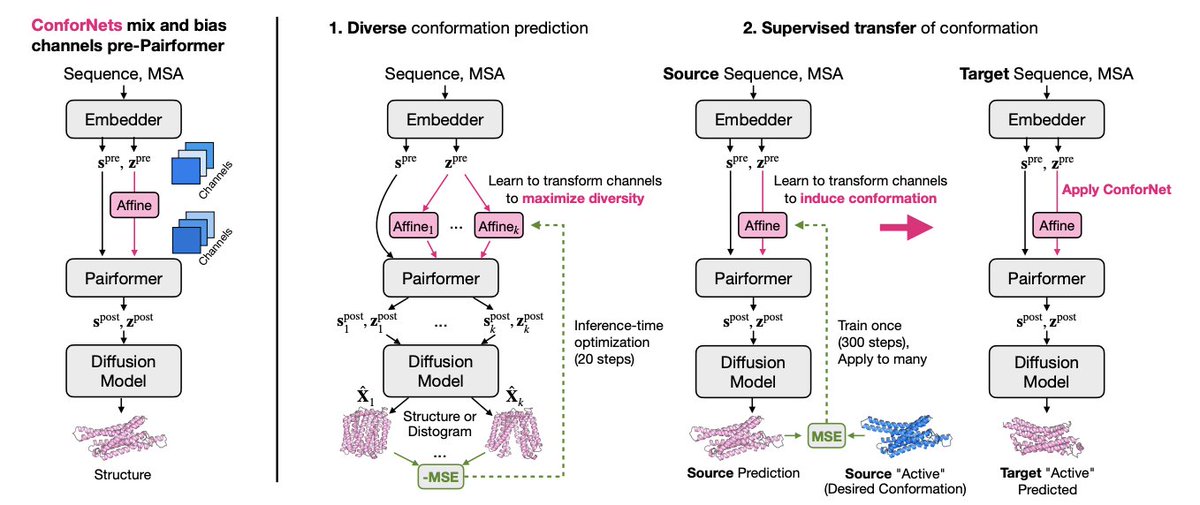
ConforNets: Latents-Based Conformational Control in OpenFold3
1. The work introduces ConforNets: lightweight, inference-time adapters for AF3/OpenFold3 that steer proteins toward alternate conformations by applying channel-wise affine transforms to the pre-Pairformer pair latents (zpre), enabling controllable conformational sampling rather than ad hoc perturbations.
2. Key design choice: instead of perturbing MSAs (combinatorial, protein-specific) or guiding diffusion directly (can be brittle), ConforNets globally modulate AF3’s internal representations upstream of diffusion, letting the diffusion module still map to a physically plausible structure manifold.
3. Mechanism: a ConforNet is an affine transform ϕ(h)=hW^T b over channels (e.g., 128 pair-latent channels), initialized as identity. Because it is channel-wise (not residue-wise), the same learned transform can be reused across proteins, including different lengths.
4. Unsupervised alternate-state generation: the method jointly optimizes multiple ConforNets for one input protein to maximize diversity between their outputs. Diversity objectives include (i) distogram CDF MSE separation and (ii) coordinate MSE separation (using a single deterministic denoising step for efficiency).
5. Efficiency note: optimization is designed to be fast and practical—reported as <40 GPU seconds for a ~200-residue protein to fit ConforNets in the diversity setting—then amortized over many diffusion samples.
6. Benchmark results (104 proteins, multiple categories: domain motions, membrane transporters, cryptic pockets, fold switchers, OOD60): ConforNets achieve the best success@100 across all benchmarks versus OF3p baselines and prior inference-time methods (e.g., AFsample3, ConforMix), with distogram-based training often strongest overall and coordinate-based training helping when strict local RMSD cutoffs dominate (cryptic pockets).
7. Qualitative behavior: despite global latent modulation, ConforNets can induce localized functional changes (e.g., cryptic pocket opening/closing) and, for transporters, sampling concentrates near the two known inward/outward basins rather than producing diffuse intermediate noise—though the resulting “energy landscapes” are explicitly not claimed to be calibrated.
8. New supervised task: “conformational transfer.” A ConforNet is trained on a single source protein with a desired reference state, then applied to other family members to induce the analogous conserved conformational change—aiming for at-will state induction rather than merely occasional sampling.
9. Transfer performance: large gains in success@5 (interpreted as controllable induction) vs default OF3p sampling—GPCR active 24%→79%, kinase DFG-out 6%→23%, transporter outward-open 16%→57%—and improvements in reachability (success@100) as well. Template-providing baselines do not replicate these improvements.
10. Analysis/ablation: the optimal intervention point is zpre (pre-Pairformer pair latents), which remains stable under full diffusion rollouts; post-Pairformer perturbations can “fit” short rollouts but degrade under full sampling. Ablations indicate that even small off-diagonal mixing in W matters; restricting to diagonal-only or removing W reduces performance, implying cross-channel interactions propagate through Pairformer updates to reshape contact reasoning.
📜Paper: arxiv.org/abs/2604.18559
#AlphaFold3 #OpenFold3 #ProteinFolding #ComputationalBiology #DeepLearning #ProteinConformations #GenerativeModels #StructuralBiology
1
4
31
2,182