
A hybrid physics-deep learning framework for combinatorial de novo design of small-molecule binding proteins
1. The paper introduces CLAIRE (CombinatoriaL Assembly with Integrated REfinement), a hybrid workflow that aims to make de novo small-molecule binder design more reliable by combining explicit physics-based interaction scaffolding with deep-learning-guided sequence/structure optimization.
2. Core idea: instead of asking generative models to “discover” atom-level protein–ligand hydrogen bonding after the fact, CLAIRE defines high-fidelity interaction motifs up front and then searches for backbones that can accommodate those motifs with tight geometric tolerances (distances within ~0.5 Å; angles/torsions within ~10–15°).
3. Motif innovation: the authors extend BSFF (Binding Sites from Fragments) by mining the PDB for residue–fragment interactions, spatially clustering them into discrete “interaction modes,” and preferentially using statistically overrepresented modes (not necessarily those best-scoring by Rosetta), capturing preferences like pi-stacking and cation–pi that energy functions may underweight.
4. Scaffold innovation: the authors generalize motif scaffolding beyond helical bundles by using LUCS to generate thousands of reshaped de novo NTF2-like alpha–beta scaffolds with finely varied pocket geometries, mimicking how nature reuses folds by subtle geometric shifts around functional sites.
5. Combinatorial matching: motifs and scaffolds are screened at scale using Rosetta Match; buried ligand placements are kept (≤30% ligand SASA exposed), yielding high matching throughput (reported as >160 buried matches per input motif across five diverse small molecules), enabling large libraries of candidate complexes.
6. Refinement step 1 (physics, targeted): HBRefine is introduced to fix a common failure mode in small-molecule binder design—buried unsatisfied polar atoms. It (a) mutates extraneous buried polar residues to hydrophobics when favorable and (b) proposes local mutations to create new H-bonds to any unsatisfied ligand polar atoms, then repacks and accepts changes if energetically non-worse.
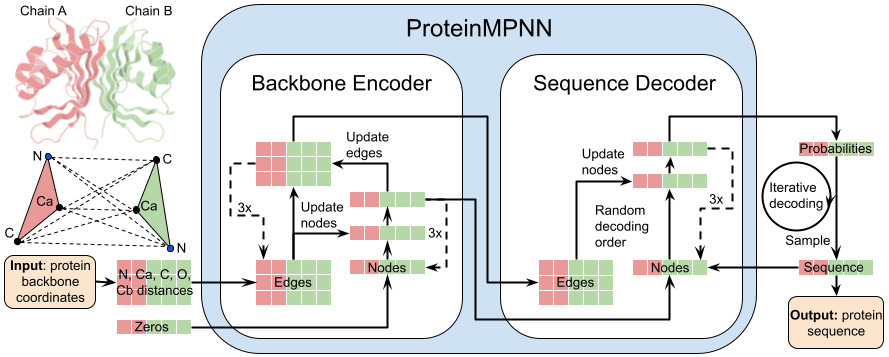
7. Refinement step 2 (ML physics): ProteinMPNN redesigns residues outside the binding site to restore global sequence–structure compatibility after pocket remodeling; Rosetta FastDesign then redesigns using MPNN-derived profiles, followed by filtering for both binding metrics (e.g., interface H-bonds, shape complementarity, ddG) and stability metrics (e.g., packstat, exposed hydrophobics, global polar satisfaction).
8. Quantitative takeaway (in silico): HBRefine plus ProteinMPNN increases the fraction of designs passing stringent multi-metric filters by up to ~7-fold. When compared to RosettaFold Diffusion all-atom pipelines on progesterone/estriol, CLAIRE yields higher in-silico pass rates; the diffusion designs most often fail on ligand H-bond satisfaction and interface buried unsatisfied polar atoms.
9. Experimental validation on two similar steroids: 26 designs (13 estriol, 13 progesterone) were tested. All expressed solubly; ~58% were monomeric by SEC; 31% were well-folded by 15N-HSQC. Binding by NMR chemical shift perturbations was observed for 1 estriol design and 3 progesterone designs, i.e., 4/26 binders overall (notably higher than typical sub-1% reports for fully generative workflows).
10. Structural and mechanistic support: NMR structures for A1E (apo/holo characterization) and D2P (holo) agree well with models (non-loop Cα RMSDs ~1.5 Å vs AF2). Motif-residue point mutations (e.g., A1E N43V/S45V/Y85F; D2P Y14F/T98V) reduce binding signals, supporting that the designed polar contacts are functionally important. Designed binding modes differ from human estrogen receptor binding solutions and show higher interaction density, indicating novelty rather than copying natural motifs.
💻Code: github.com/cvgalvin/CLAIRE
📜Paper: biorxiv.org/content/10.64898…
#ProteinDesign #ComputationalBiology #Rosetta #ProteinMPNN #AlphaFold2 #NMR #DeNovoDesign #SmallMoleculeBinding #HybridModels #StructuralBiology

1
6
37
2,517

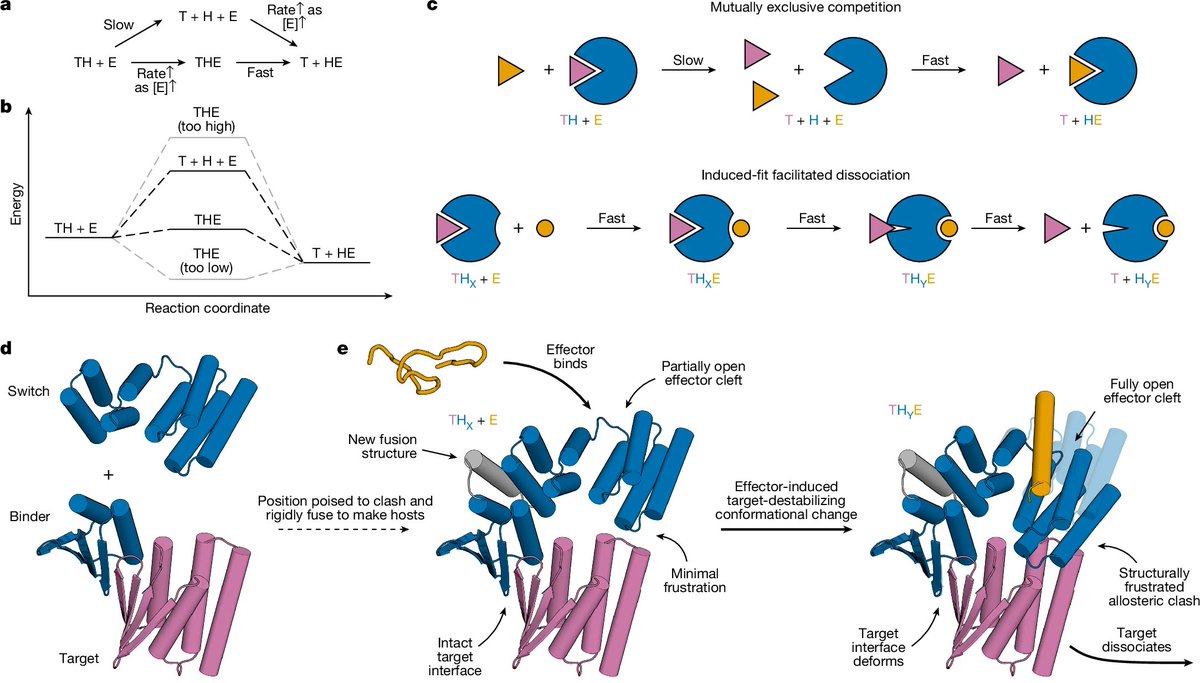
Design of facilitated dissociation enables timing of cytokine signalling | @Nature
- Design a general protein strategy that controls dissociation kinetics of protein complexes by enabling facilitated dissociation through effector-responsive conformational switches
- Construct switch–binder fusions by first sketching placements in PyMOL (no clash in state X, strong clash in state Y). Generate fusion backbones with RosettaFold inpainting (early designs) or Rosetta FastDesign RFdiffusion (later designs), masking noncritical residues at fusion interfaces. Optimize sequences using ProteinMPNN for both switch and binder regions
- Predict structural states with AlphaFold2 (AF2, AF2-IG) for host alone, host target, and host effector. Model strained ternary intermediates with AF2-IG to ensure diverse deformation directions and favorable strain distribution
- Demonstrate up to 5,700-fold acceleration of dissociation by SPR, confirm designed clashes and conformational states with X-ray crystallography, and show rapid, tunable control in applications
Link: nature.com/articles/s41586-0…
6
8,159

28 Jul 2025
Hurry up and get early access to figma design sections.
don't design from scratch, create from ready-made premium sections from professional designers.
uiblockify.co
#uiblockify #design #figmadesign #uiux #saas #landing #designers #fastdesign #startup #madeinfigma
1
2
43
8 Jul 2025
Speed up your design workflow.
Get early access to a growing library of ready-to-use UI sections for your next landing or project.
#figma #design #fastdesign #tools #saas #Website #uiuxdesign
2
49

29 Jun 2025
From idea to identity. In minutes.
That's not just our tagline; it's our promise. Watch how Brandiseer delivers a stunning, launch-ready brand kit at the speed of your ambition.
#Brandiseer #IdeaToLaunch #FastDesign #AIRevolution
2
53

15 Mar 2025
🔥 Step into the DESIGN RING and create pro-level mockups in SECONDS—no design skills needed! 🎨💪 Use lightning-fast templates, customize every detail, or upload your own artwork to make designs that OOZE CHAMPIONSHIP GOLD! 🏆 🚀 #MockupMadness #DesignLikeAChamp #MachoMockups #GraphicDesign #DesignTools #CreativePower #MockupGenerator #ProDesigns #EasyMockups #DesignMadeSimple #CustomMockups #Branding #EntrepreneurLife #PrintOnDemand #MarketingDesign #CreativeBusiness #LogoDesign #TShirtDesign #VisualBranding #FastDesign #ai #aifree #aibusiness
1
43

11 Mar 2025
Transform Your Brand with a Luxury Logo in Just 12 Hours!
A modern, minimalist, and luxurious logo is the perfect way to elevate your brand's image. We deliver high-quality logos within 12 hours
#BrandIdentity #LuxuryLogoDesign #MinimalistLogo #LogoDesign #FastDesign

1
2
53

25 Jan 2025
Sonicxswap makes DeFi easy with its simple, fast, and user-friendly design. @Sonicx_Dex
Website: sonicxswap.com/
All links: linktr.ee/sonicxswap
#Simplicity #FastDesign #UserFriendly #Sonicxswap
#Blockchain

9
54
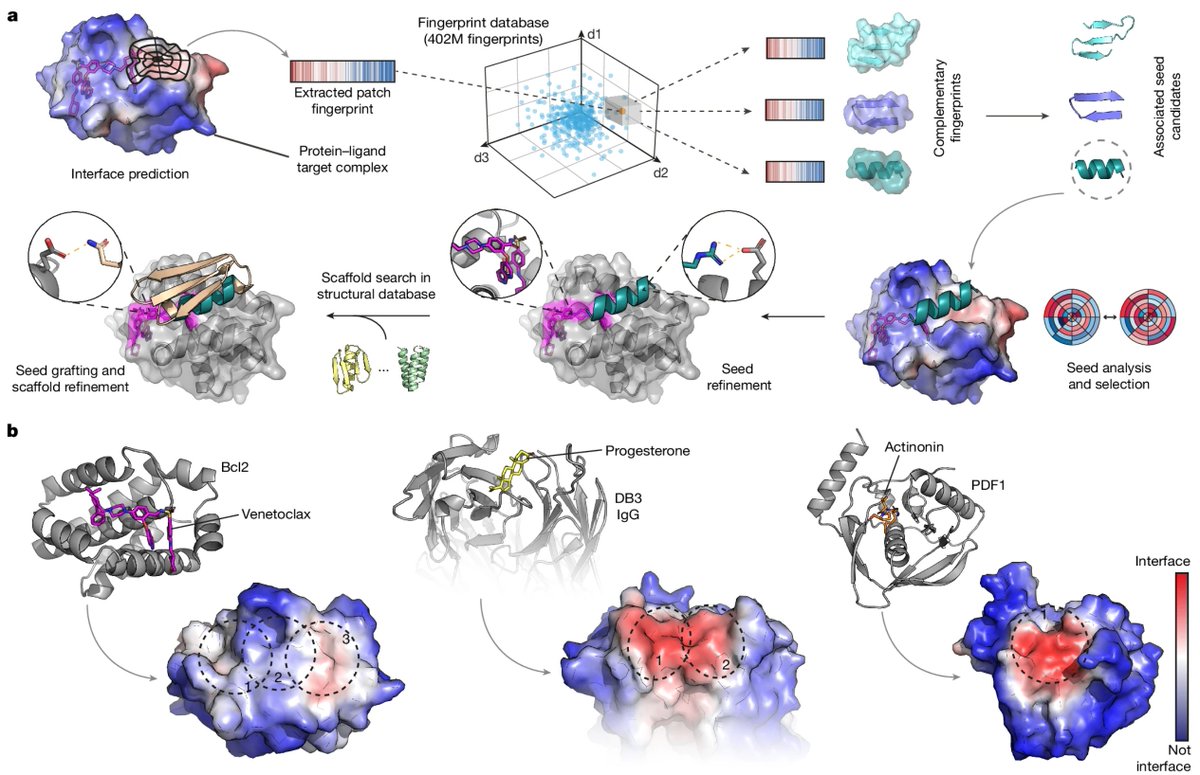
Targeting protein–ligand neosurfaces with a generalizable deep learning tool | @Nature
- MaSIF-neosurf can design binders for protein-ligand complexes, targeting neosurfaces (i.e., ligand-induced structural changes on the protein surface)
- Benchmark MaSIF-neosurf against RFAA on 14 ligand-induced PPI complexes with 8,907 decoys from PDBBind
- Use MaSIF-search to predict buried surfaces and identify complementary surface fingerprints from a database of protein fragments (~640,000)
- Construct full-length proteins with binding motifs and refining structures using the Rosetta FastDesign protocol and grafting (with a potential round of LigandMPNN optimization)
- Engineer and validate binders for Bcl2–venetoclax, DB3–progesterone, and PDF1–actinonin through experimental testing
Link: nature.com/articles/s41586-0…
19
36
3,231

19 Oct 2024
Need a logo or website fast? ⚡️ OneDayDesignGroup.com delivers pro logo design and web design in just 24 hours! 🚀 Perfect for startups, rebrands, or quick projects. Get high-quality design, fast turnaround, and zero hassle! 🎨💻 #LogoDesign #WebDesign #OneDayDesign #FastDesign
1
2
41
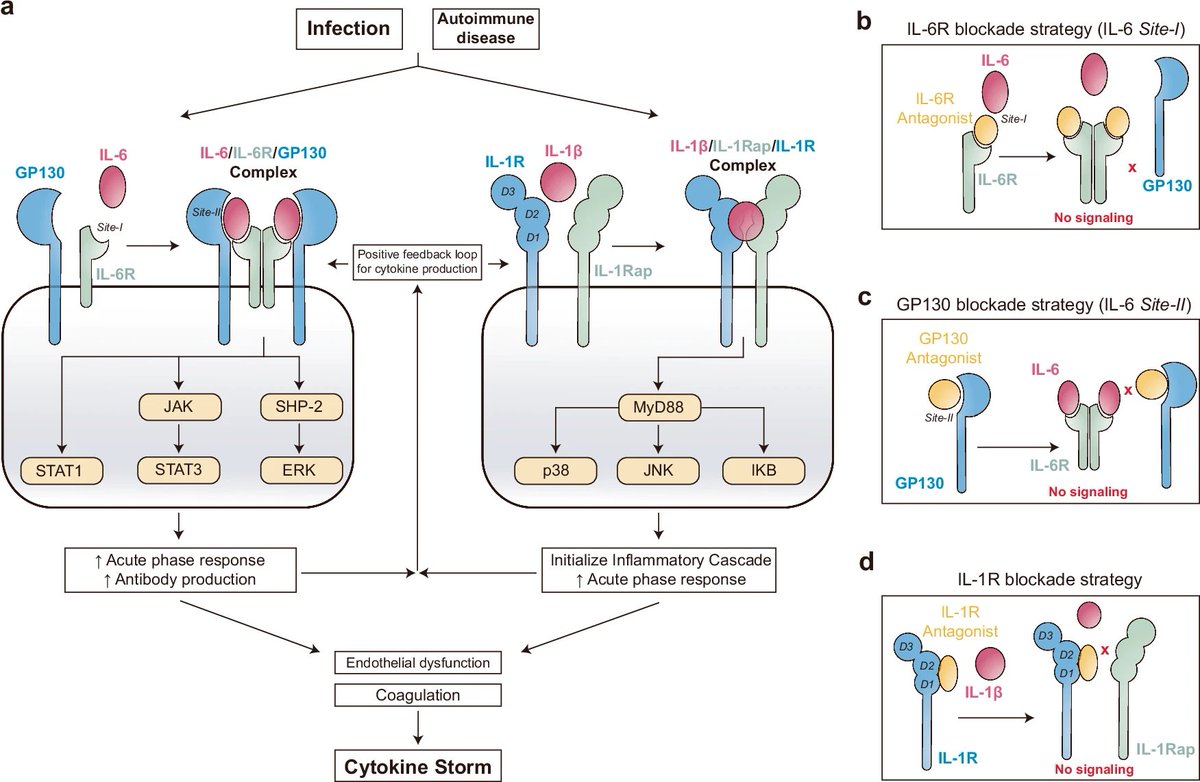
De novo design of miniprotein antagonists of cytokine storm inducers | @NatureComms
- Use Rosetta-based binder design approach (Cao, et al., 2024) against IL-6R, GP130, and IL-1R1
- Dock 40k de novo protein scaffolds to hotspot residues (Patchdock and Rifdock), and design 2.5 million backbones to sequences (Rosetta Fastdesign)
- Filter top designs based on Rosetta metrics and screen the best 100k candidates using yeast display
Link: nature.com/articles/s41467-0…
17
84
4,434

15 Aug 2024
Everyone studying molecular biology should track how other researchers connect AI models into relevant workflows.
Here's an example for protein assembly design:
De novo design of allosterically switchable protein assemblies
nature.com/articles/s41586-0…
Models: ProteinMPNN, RFDiffusion, AlphaFold2 -multimer, HFuse, Rosetta FastDesign, WORMS, RPXDock46
2
5
329

12 Aug 2024
Infinite Designs with Marvelloux UIUX Studio's Pixelplan!
Introducing the Pixelplan – the ultimate subscription-based service tailored to meet all your design needs! Join today and gain instant access to your personalized Trello board, where you can assign us an unlimited number of tasks.
Why Pixelplan is Perfect for You:
🚀 Flexibility at Your Fingertips: Customize your design requirements with ease.
⚡ Rocket-Fast Delivery: Get your designs done quickly without compromising on quality.
🔄 Unlimited Designs Revisions: No limits, just endless creativity and improvements.
🎨 Expert Designs: Our team of seasoned professionals is here to bring your vision to life.
💰 Value for Money: Get top-notch designs without breaking the bank.
📈 24/7 Progress Tracking: Stay updated on your projects round the clock.
Ready to elevate your brand with limitless designs? Join now and experience the power of the Pixelplan – where creativity knows no bounds!
For more details contact us here website : pixelplan.co/
#UnlimitedDesigns #GraphicDesign #UIUX #PixelPlan #CreativeServices #DesignSubscription #FastDesign #DesignStudio #ExpertDesigns #BrandDesign #MarketingDesign #WebDesign #DesignSolutions #DesignAgency #MarvellouxStudio
1
1
3
69


3 Aug 2023
Foldit standalone上でRosettaScriptを使ってFastDesignしたgif。movemapでbbをFalseにしているので、バックボーンを固定してロータマー探索している動画になっているはず。
1
6
576

6 May 2023
AI and the Future of Design
#aidesign #designbyai #Ai #artificialintelligence #automateddesign #designauto #designerAi #Aidesigner #designwithAi #chatgtpforDesigner #fastdesign #designergrowth




1
43

14 Feb 2023
Learned something new today.
Create a new Design System - Library of Components integrating with Angular, React, Svelte, Vue, etc,. using one & Only tool - fast.design/
#fast #fastdesign @Microsoft #microsoft #javascript #js #angular #react
1
2
58

15 Dec 2022
تصميم سوشيال ميديا من سلسلة تصاميم في السريع
#socialmediadesign
#combo #fastdesign #photoshop #ilustration #mydesign
@Designers_Sup @Des_together @YOUR_PLATFORMM @OCreativeJobs @Scheme125 @wesllahh @dam_designers
#تجمع_فنانيين٩ #مصممين

1
4
4

16 Sep 2022
Pipeline updated! ProteinMPNN is great helpful in providing better design after FastDesign, thank all contributors!
15 Sep 2022

ProteinMPNN paper: science.org/doi/10.1126/scie…
Github: github.com/dauparas/ProteinM…
Colab notebook: github.com/dauparas/ProteinM…
HuggingFace notebook: huggingface.co/spaces/simond…
Hallucinating symmetric protein assemblies paper:
science.org/doi/10.1126/scie…
Thanks to all collaborators!
2
26 May 2022
FastDesign(FastRelax)を使う場合は、オプションで渡すことができるresfileでNATAAにして、かつ、MoveMapのbbを0に指定すると、側鎖を変えずに側鎖のrepackだけ行われて主鎖は動かないと思います。複数のchainがあって、その主鎖の相対的な位置を動かさずにするには、MoveMapでjumpを0にします。
2
2