
Jun 10
HOMO-LUMO gap tells you a lot about a molecule's reactivity before you ever run a reaction!
A small gap = chemically reactive, good for catalysis but potentially unstable as a drug candidate. A large gap = kinetically stable, better for therapeutic applications. The gap value directly informs electrophilicity, nucleophilicity, and selectivity predictions.
Our DFT module computes HOMO-LUMO descriptors at near-SOTA accuracy (mean gap: 4.16 eV, 10/10 QC checks passed) — reliable across non-aromatic systems, with GPU4PySCF acceleration.
Physics-informed molecular intelligence. Not just data-driven guessing.
Explore at boltzmann.co
#HOMOLUMO #DFT #QuantumChemistry #ComputationalChemistry #MolecularIntelligence #DrugDesign #AIChemistry
3
33

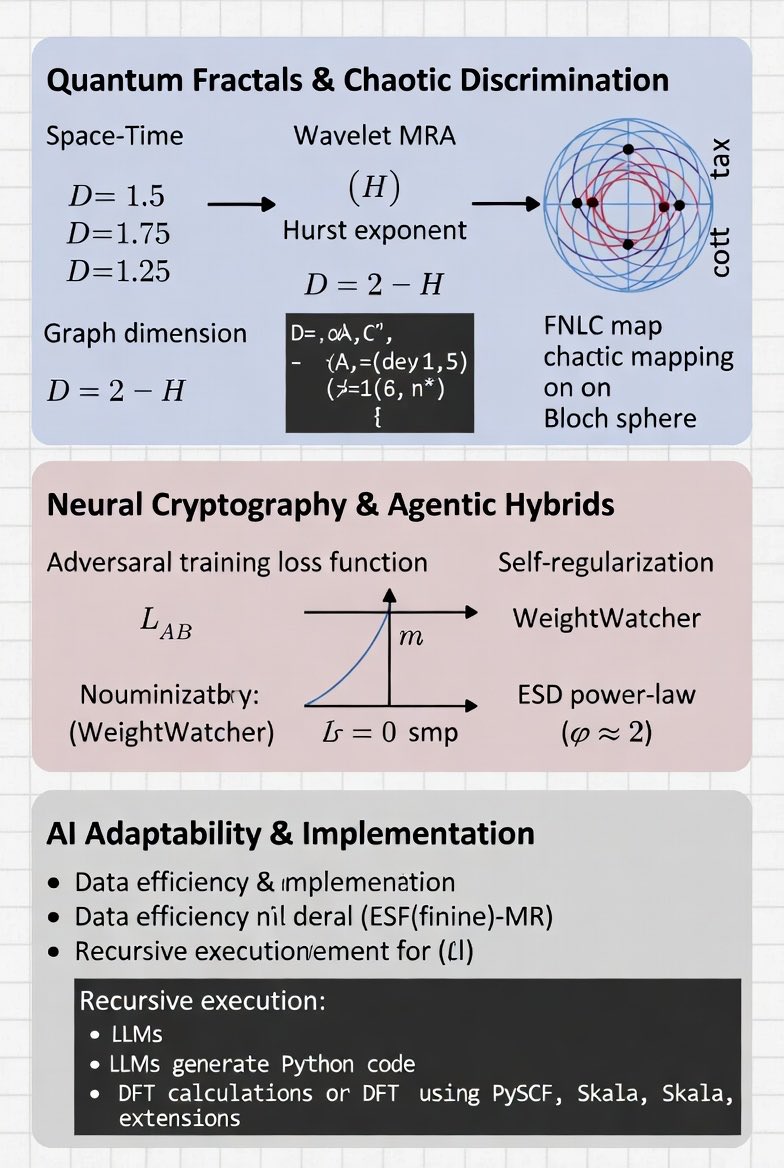
Quantum Fractals & Chaotic Discrimination (Adaptability in Dynamics)
Infinite square-well carpets: space fractal dimension (D_space = 3/2), time (D_time = 7/4), space-time (D_space-time = 5/4) (Berry). Wavelet MRA: detail energies (E_j ∼ 2^{-jα}), Hurst (H = -α), graph dimension
[D = 2 - H.]
Chaos-mediated discrimination (FNLC map on Bloch sphere):
[f(z) = (2sz 1)/(2z s), s = i]
(yields exponential divergence; Leggett-Garg correlation (r_XY → 0) after waiting time τ(δ)).
Neural Cryptography & Agentic Hybrids (Direct AI/LLM Tie-In)
Adversarial training losses:
[L_B = 1/N Σ |P_i - B(C,K)_i|, L_E = E[L1(P, E(C))], L_AB = L_B - λ L_E.]
SETOL/HTSR monitors ESD power-law (ρ(λ) ∼ λ^{-α}) (ideal α ≈ 2); probit uncertainty (P(y=1|x) = Φ(x^T β)). Recursive LLMs enable executable code generation for state inspection/transforms.
Programming AI Adaptability for Agents and LLMs
Parrish’s threads advocate hybrid, data-efficient programming for adaptability—echoing Skala’s two-stage training and Agency Efficiency Principle (quality workflows > raw scale). Agents/LLMs become “adaptable” via recursive/self-referential loops, tool-use (TPTU-style), fidelity-aware scheduling, and quantum-classical orchestration. Best practices: curate ~78 high-quality full-workflow examples (beats 10k synthetics); monitor via WeightWatcher for self-regularization; integrate quantum simulators (PySCF Skala) for chemistry agents.
Practical Implementation Sketch (Python/PyTorch Skala/PySCF Extensions)
# Hybrid Agentic LLM Skala-DFT for Scientific Adaptability
import torch
from pyscf import gto, scf
from skala.pyscf import SkalaKS
import weightwatcher as ww
# 1. Recursive LLM Agent (adaptability via code-gen self-correction)
class RecursiveAgent:
def __init__(self, llm):
self.llm = llm
self.history = []
def plan_and_execute(self, task, quantum_mol=None):
code_prompt = f"Generate PySCF Skala code for {task} with error correction."
plan = self.llm.generate(code_prompt)
try:
exec(plan)
mol = gto.M(atom=quantum_mol, basis="def2-tzvp")
ks = SkalaKS(mol, xc="skala-1.1")
e = ks.kernel()
self.history.append(e)
except Exception as err:
correction = self.adapt_via_weightwatcher(plan)
return self.plan_and_execute(task, quantum_mol)
return e
def adapt_via_weightwatcher(self, model_code):
watcher = ww.WeightWatcher(model=eval(model_code))
df = watcher.analyze(detX=True)
if df['alpha'].mean() < 1.8:
return "adjusted plan with α→2 regularization"
return model_code
# 2. QHPC Orchestration
# Fidelity-aware scheduler pseudocode:
# score = fidelity * (1 - latency_µs/4) * parallelism_factor
Adaptability Best Practices (Parrish-Style)
• Data Efficiency: 78 curated workflows > 10k samples.
• Monitoring: WeightWatcher probit heads for uncertainty; enforce ERG (det X ≈ 0).
• Hybrid Scaling: Embed Skala DFT calls in LLM agents for real-time molecular QM (reaction energies at hybrid accuracy, O(N) cost).
• Error Mitigation: Chaotic amplification Leggett-Garg witnesses; photonic cooling Lindbladian for hardware-in-loop agents.
• Extensions: Fork Skala GitHub GPU4PySCF; add recursive loops for long-horizon agentic behavior.
This framework—drawn directly from @kparrish51’s threads—makes AI agents/LLMs systematically improvable for quantum chemistry, materials discovery, and beyond. Parrish’s X threads serve as living, equation-rich reviews.


1
6
5
140

UBio-MolFM: A Universal Molecular Foundation Model for Bio-Systems
1. UBio-MolFM targets the “scale–accuracy gap” in biomolecular simulation: it aims to deliver ab initio-like DFT fidelity while scaling to solvated, heterogeneous bio-systems that are too large for standard QM and too complex for fixed-form classical force fields.
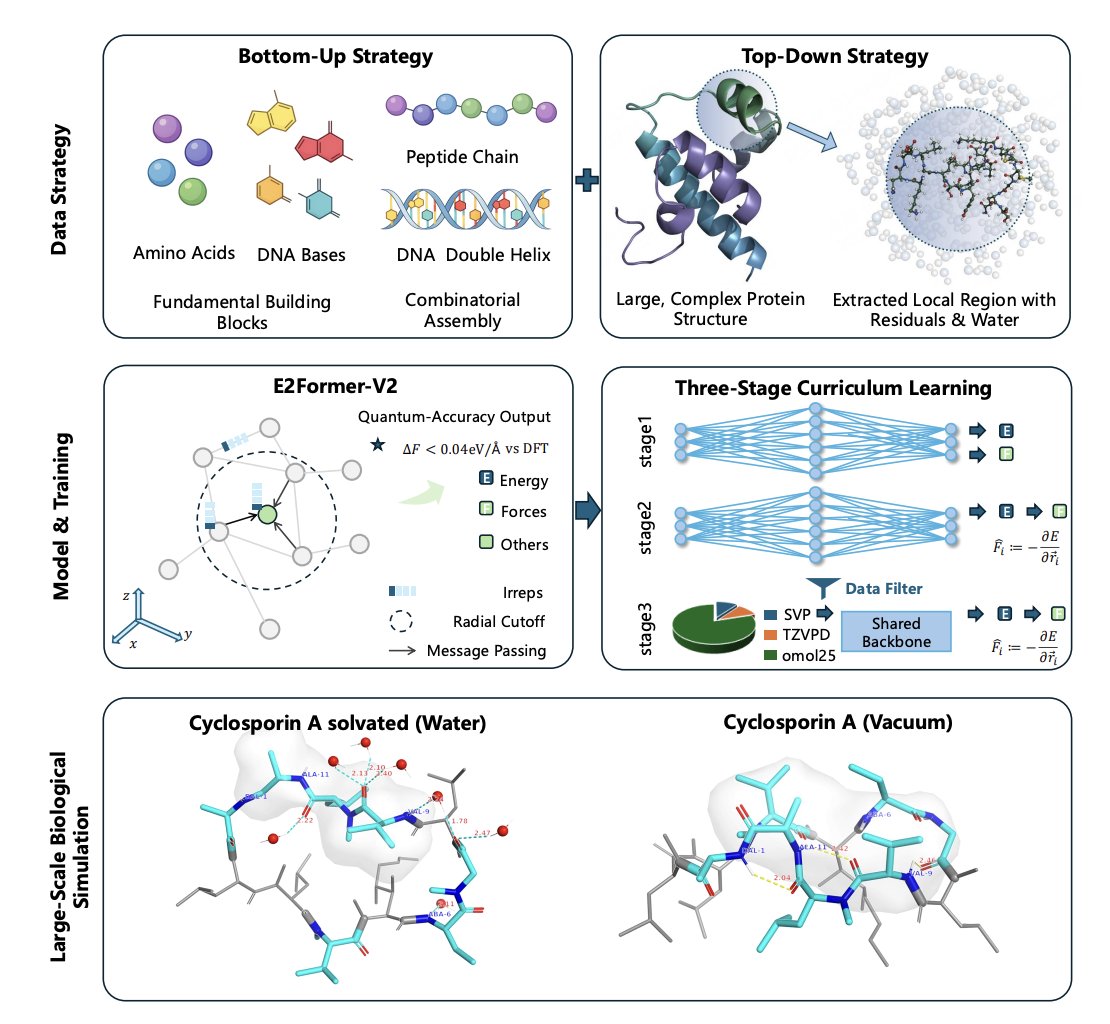
2. The framework is built on three coupled pillars: (i) UBio-Mol26, a bio-specific multi-fidelity dataset up to ~1,200 atoms; (ii) E2Former-V2, a linear-scaling SO(3)-equivariant transformer optimized for large systems; (iii) a three-stage curriculum that enforces energy–force consistency and handles heterogeneous reference levels.
3. Data innovation: UBio-Mol26 contains ~17M configurations and is designed explicitly for biology (proteins, nucleic acids, lipids, drug-like molecules, and cross-modal interactions) in explicit solvent, with element coverage including biologically important ions/metals. It complements small-molecule-centric datasets by emphasizing macromolecular chemical environments (e.g., enriched amide and methylene motifs).
4. A “Two-Pronged Strategy” is used to build UBio-Mol26: bottom-up enumeration of biochemical building blocks (e.g., exhaustive tripeptides) plus top-down sampling of native protein environments by extracting residue-centered solvated clusters from AlphaFold DB structures, with chemical capping to preserve realism.
5. Multi-fidelity QM labeling is treated as a first-class design choice to make large-system DFT feasible: ωB97M-D3 is used (cost-reduced vs VV10 variants), a mixed basis strategy improves SCF convergence, and a large def2-SVP subset enables ~10× more data at a small fraction of compute, while retaining a higher-fidelity def2-TZVPD subset.
6. Architecture innovation: E2Former-V2 combines (a) node-centric factorization to reduce edge materialization, (b) Long–Short Range (LSR) modeling to capture non-local physics without fully connected atomic graphs, and (c) Equivariant Axis-Aligned Sparsification (EAAS) that reduces dense SO(3) tensor products into sparse operations via axis-aligned frames while preserving exact equivariance.
7. Systems-level innovation: a fused “on-the-fly” equivariant attention kernel (Triton) computes attention with online softmax and streaming reductions, avoiding storing per-edge attention tensors. This is positioned as a practical route to improved memory locality and throughput on large atom counts.
8. Training innovation: a Three-Stage Curriculum Learning protocol: Stage 1 initializes on OMol25 with separate energy/force heads (fast, avoids autograd forces); Stage 2 enforces conservative forces via F = −∇E; Stage 3 mixes OMol25 with UBio-Mol26 using dual heads (SVP vs high-fidelity), dataset balancing (8:1:1), filtering for compatibility, and force-only supervision for subsets with systematic energy offsets.
9. Large-system OOD generalization is explicitly tested beyond the training size cap: test systems are ~1,300–1,500 atoms (proteins/DNA/RNA optimization and solvated-protein MD clusters), with DFT references computed using GPU4PySCF. UBio-MolFM Stage 3 substantially improves protein and RNA force/relative-energy accuracy versus general-purpose baselines (MACE-OMol, UMA-S-1p1), while highlighting a remaining weakness: DNA temporal energy stability (∆E) can regress, motivating targeted DNA data expansion.
10. Downstream MD fidelity is evaluated on macroscopic/structural observables: liquid water O–O RDF matches experimental structure; 0.15 M NaCl shows realistic ion hydration peaks and coordination numbers; Cyclosporine A maintains solvent-dependent open (water) vs closed (vacuum) conformations via H-bond competition; RNA (1L2X) Mg2 binding geometry is reproduced with more realistic Mg–O and Mg–O–P distributions than Amber99 OL3 and the tested ML baseline.
11. Efficiency results (single H100, conservative-force setting): UBio-MolFM (S3, 24M) reports markedly higher throughput on large systems (e.g., 1K atoms: 61 steps/s vs UMA-S 16; 10K: 6.1 vs 1.6; 50K: 0.72 vs 0.20), while noting memory limits when long-range interactions are enabled at extreme sizes (100K atoms OOM for UBio-MolFM in the reported setting).
12. Release plan and resources: authors describe an open-science release including pretrained weights, an inference engine, and a representative dataset subset. A public protein-focused subset (UBio-Protein26 5M) is provided for benchmarking, alongside code and model checkpoints intended to lower barriers for QM-accurate biomolecular simulation workflows.
💻Code: github.com/IQuestLab/UBio-Mo…
📜Paper: arxiv.org/abs/2602.17709
#ComputationalBiology #MolecularDynamics #MachineLearning #EquivariantNetworks #ForceFields #QuantumChemistry #FoundationModels #ProteinSimulations #RNADynamics #ScientificMachineLearning
3
7
38
3,519

Mar 27
#キャルちゃんのquantphチェック
Kohn-Shamミサイル種汎関数理論計算において、Fock buildsと核勾配の効率的な評価を実現するための、GPUアクセラレーションマルチグリッドガウシアン平面波密度フィッティングの提案。これをPySCFのGPU4PySCFに実装した。
arxiv.org/abs/2603.24881
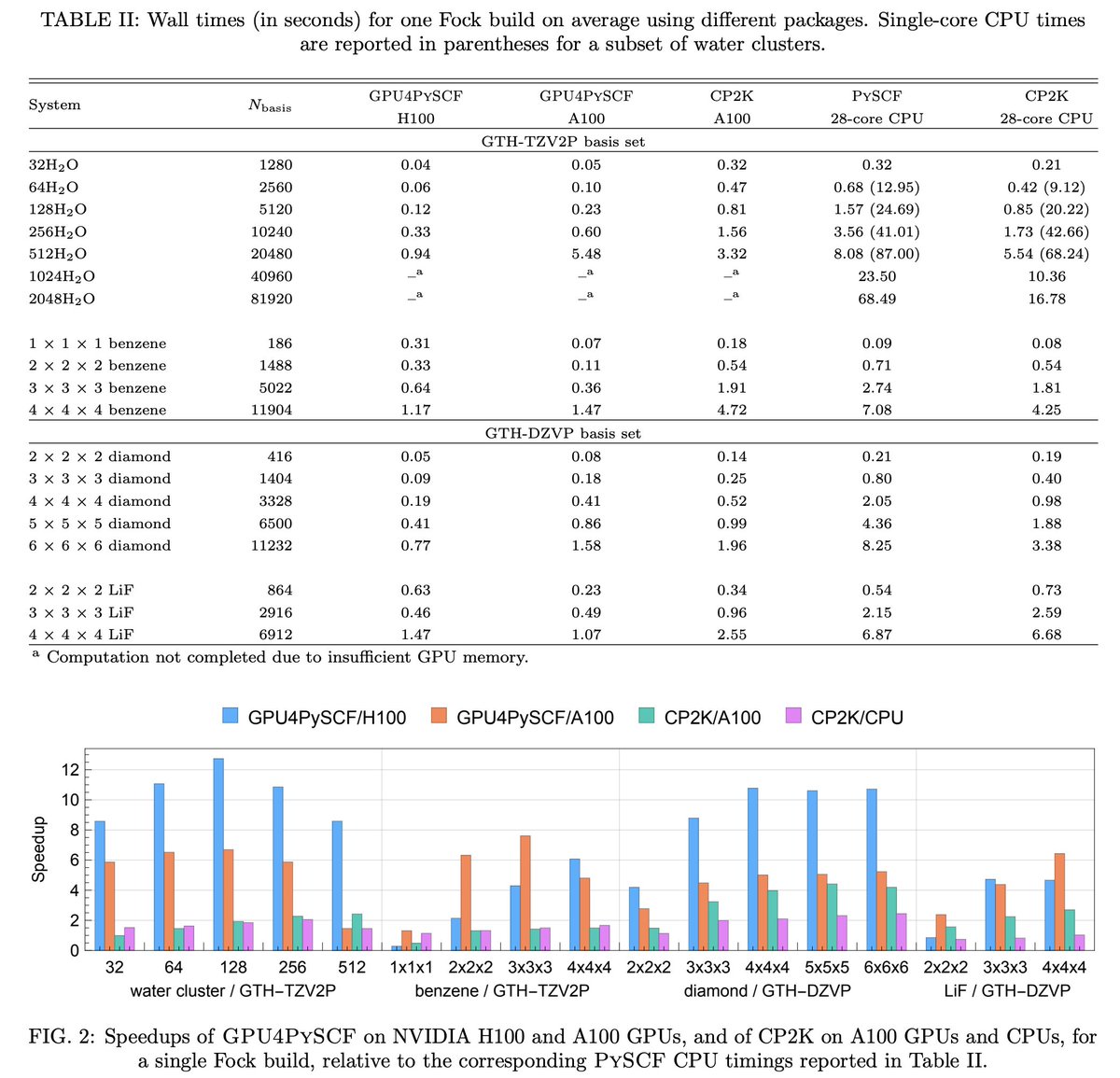
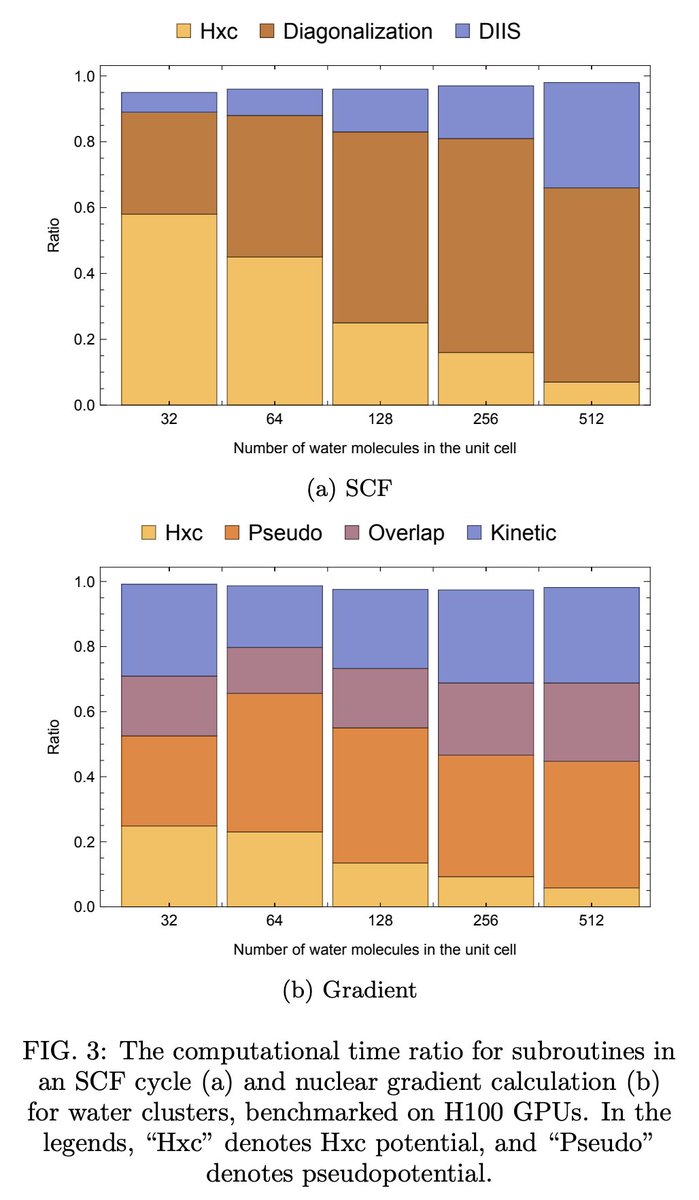
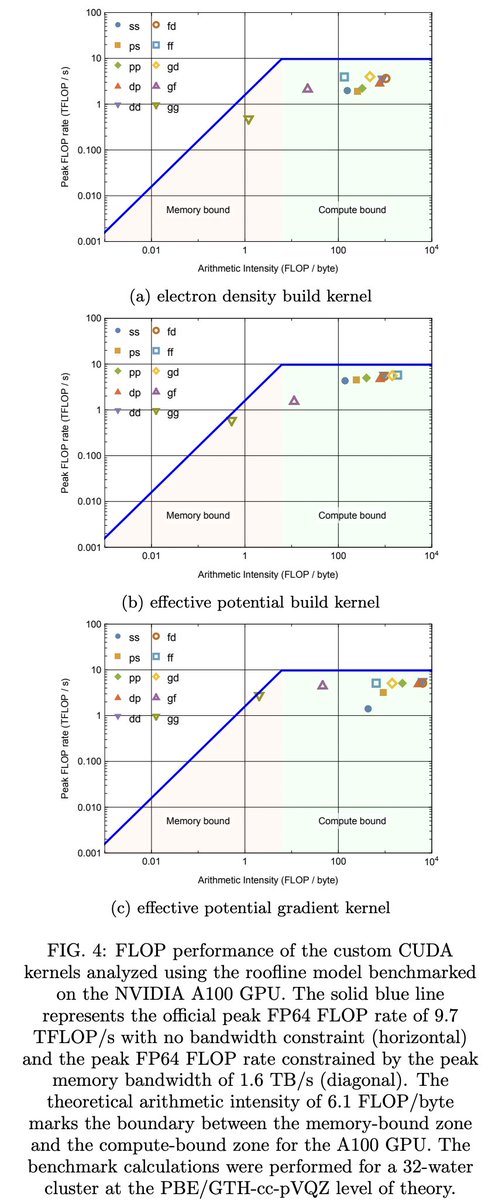
1
1
3
758

Skala community edition update: GPU4PySCF support is in ✅
That means Skala can plug into GPU-accelerated PySCF DFT workflows via GPU4PySCF (API-compatible with PySCF).
Install docs are in the repo README: github.com/microsoft/skala
#DFT #PySCF #GPU #GPU4PySCF #CompChem
3
5
1,742

19 Nov 2025
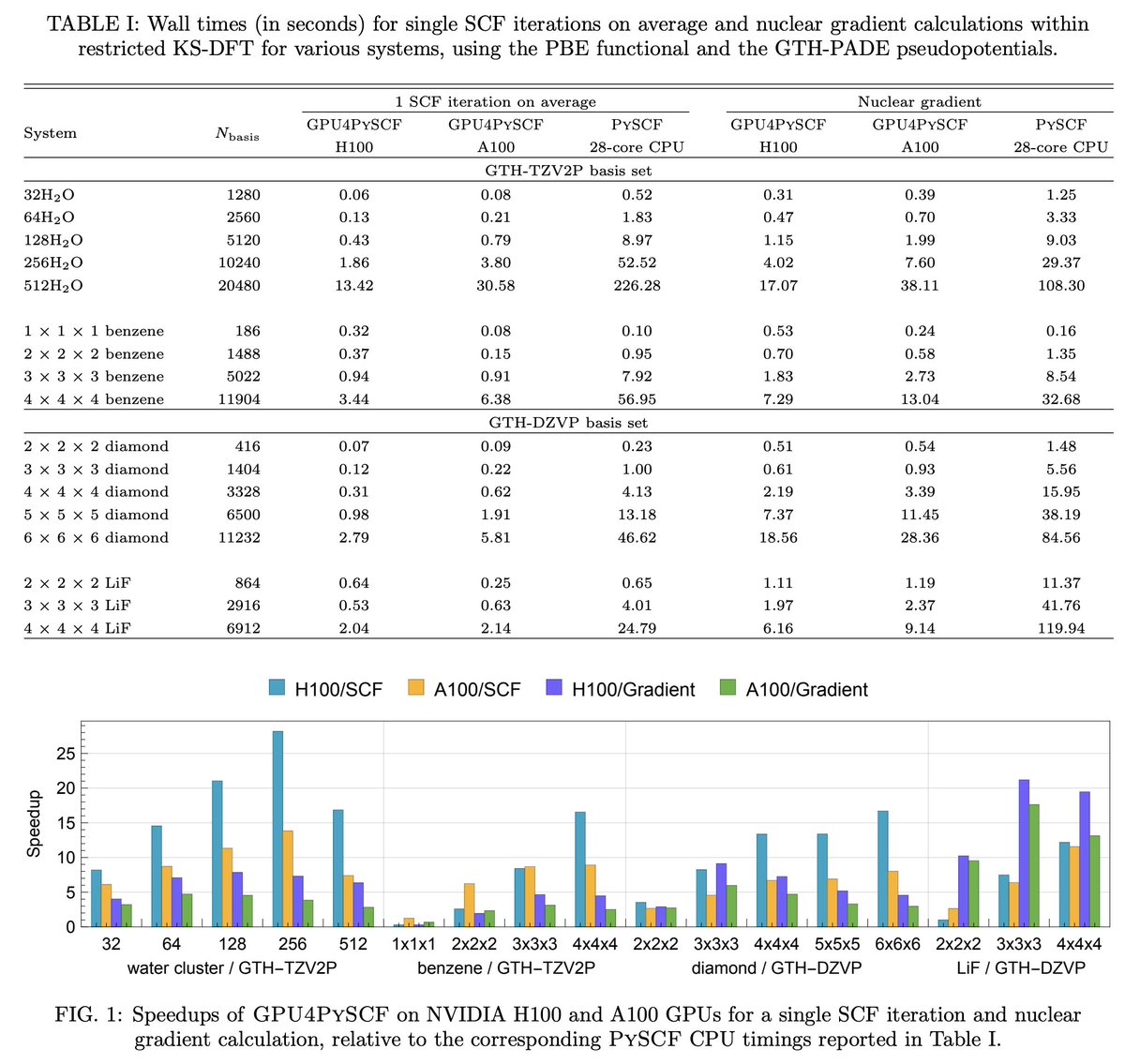
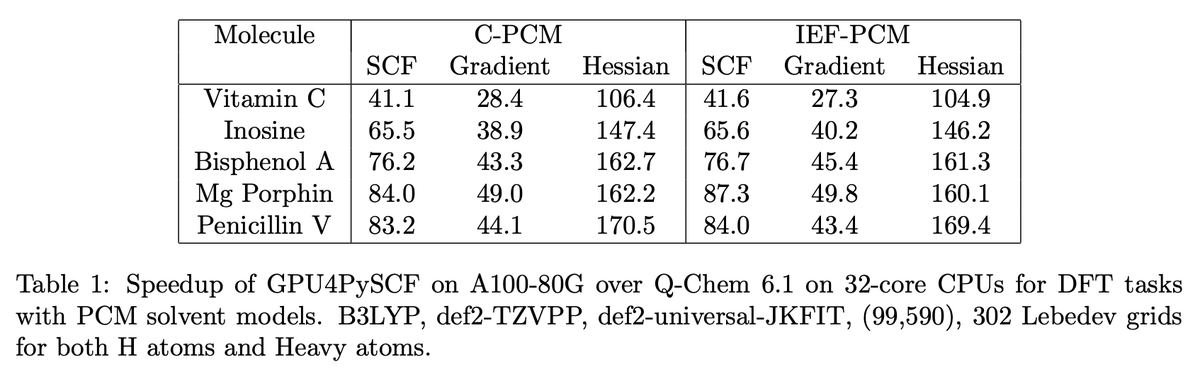
GPU4PySCF is also cost-effective. Despite the fact that the H200 instance used above costs $5–10/hr (depending on cloud provider), GPU4PySCF is so fast that it's much cheaper than running DFT on CPU codes.
Many more graphs in Jonathon's technical blog: rowansci.com/blog/gpu4pyscf
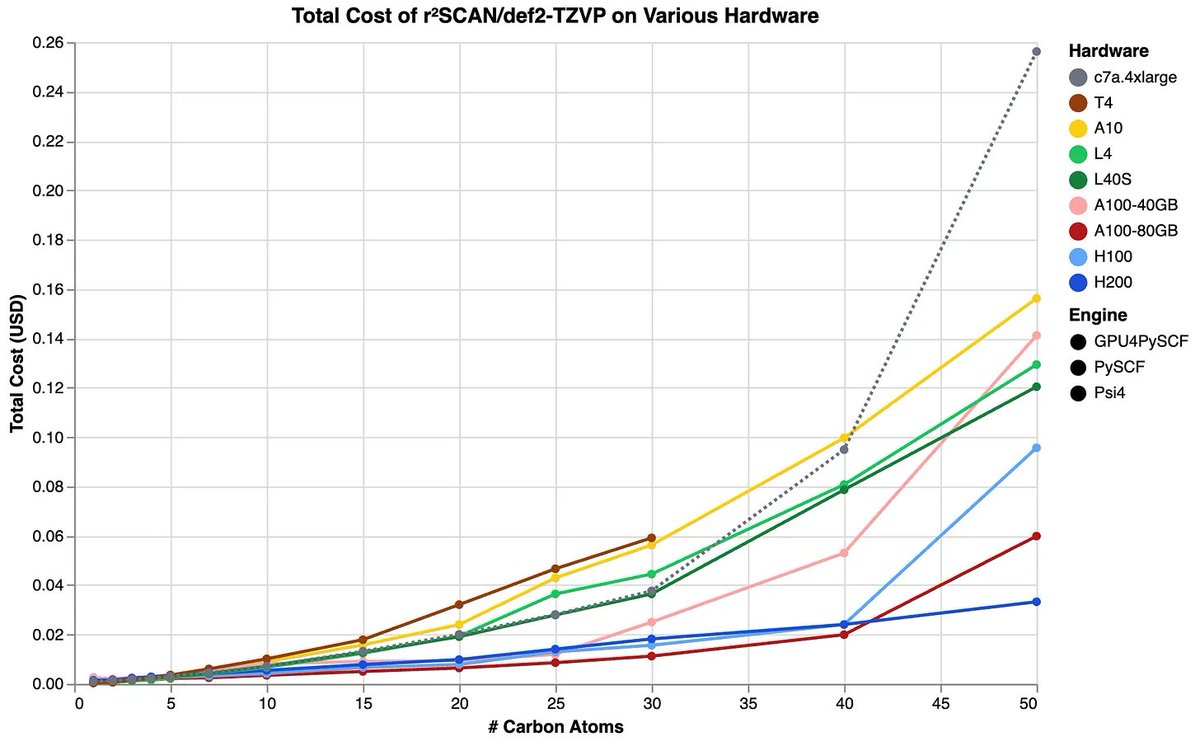
2
1
7
485
19 Nov 2025
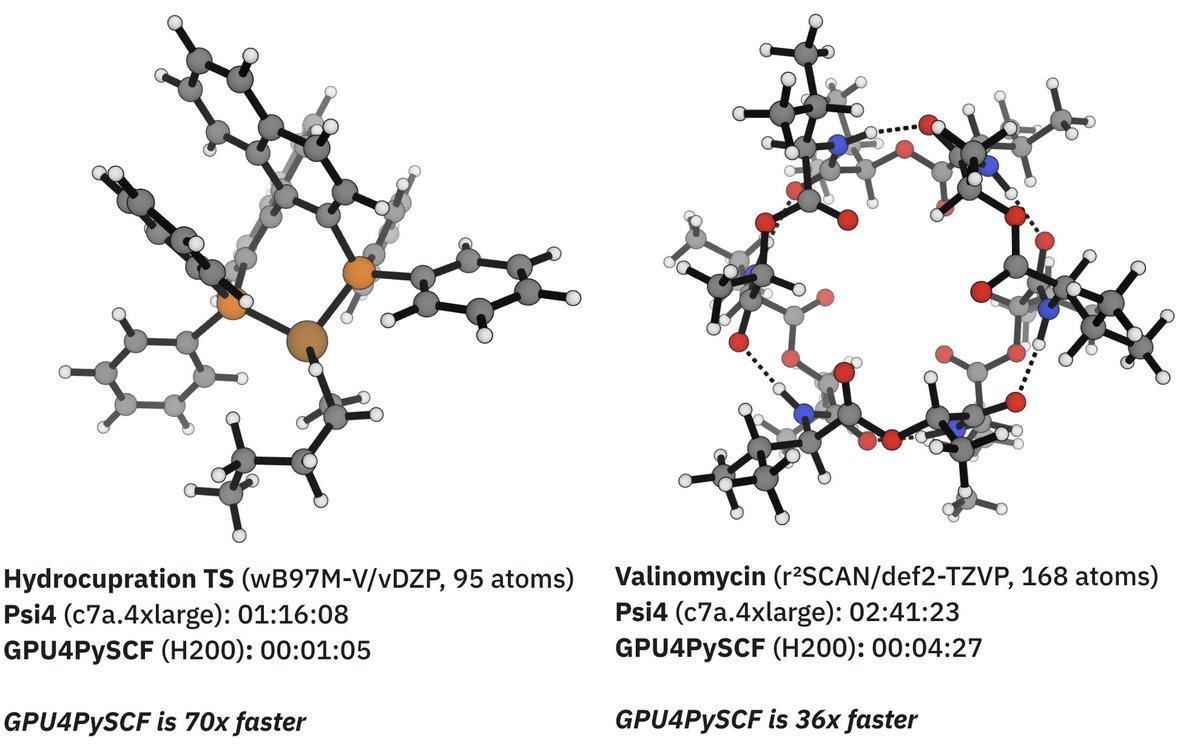
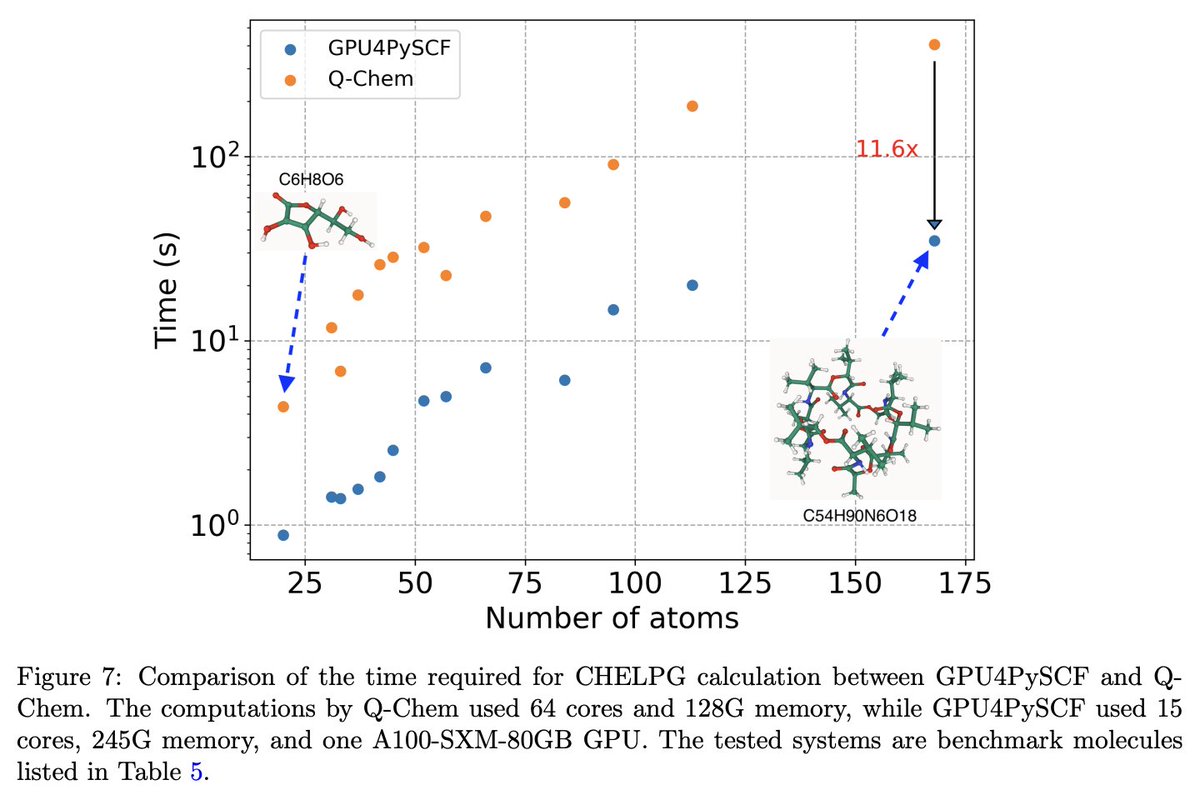
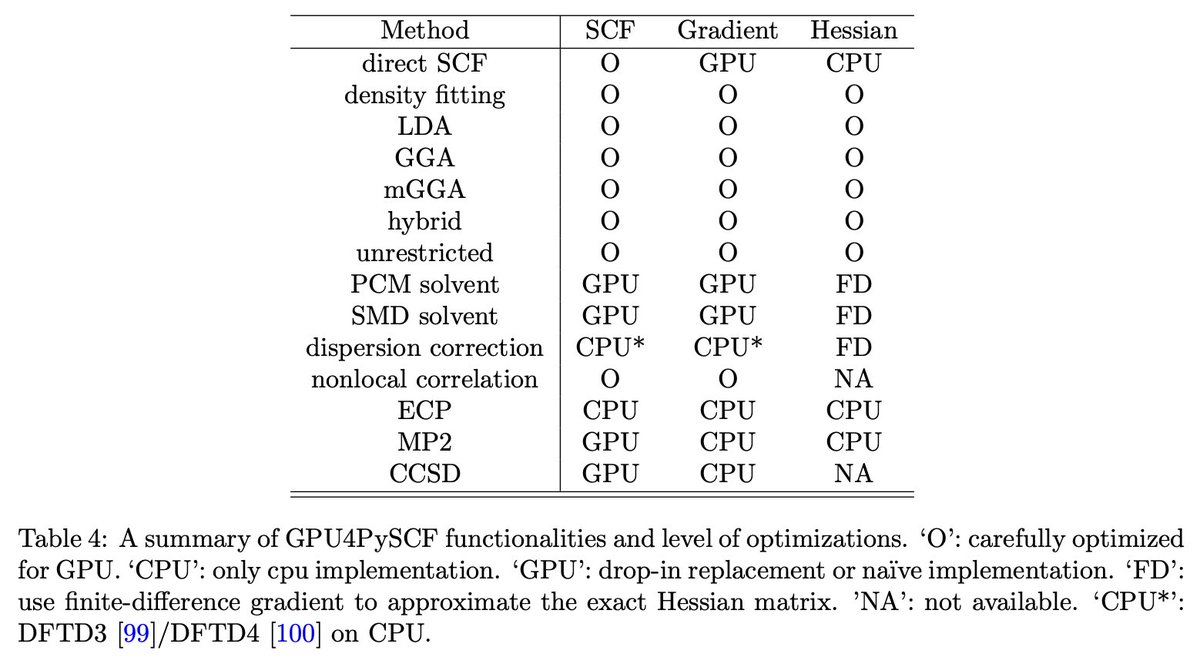
Today, we're launching GPU-accelerated DFT for all Rowan users.
Using GPU4PySCF, we see massive speedups for routine DFT tasks, making it significantly cheaper and faster to run high-accuracy quantum chemistry through Rowan.
Here's two representative examples:
7
14
102
6,855

16 Jan 2025
#compchem Good read: Accurate QM/MM Molecular Dynamics for Periodic Systems in GPU4PySCF with Applications to Enzyme Catalysis pubs.acs.org/doi/10.1021/acs…
10
53
2,367

16 Jul 2024
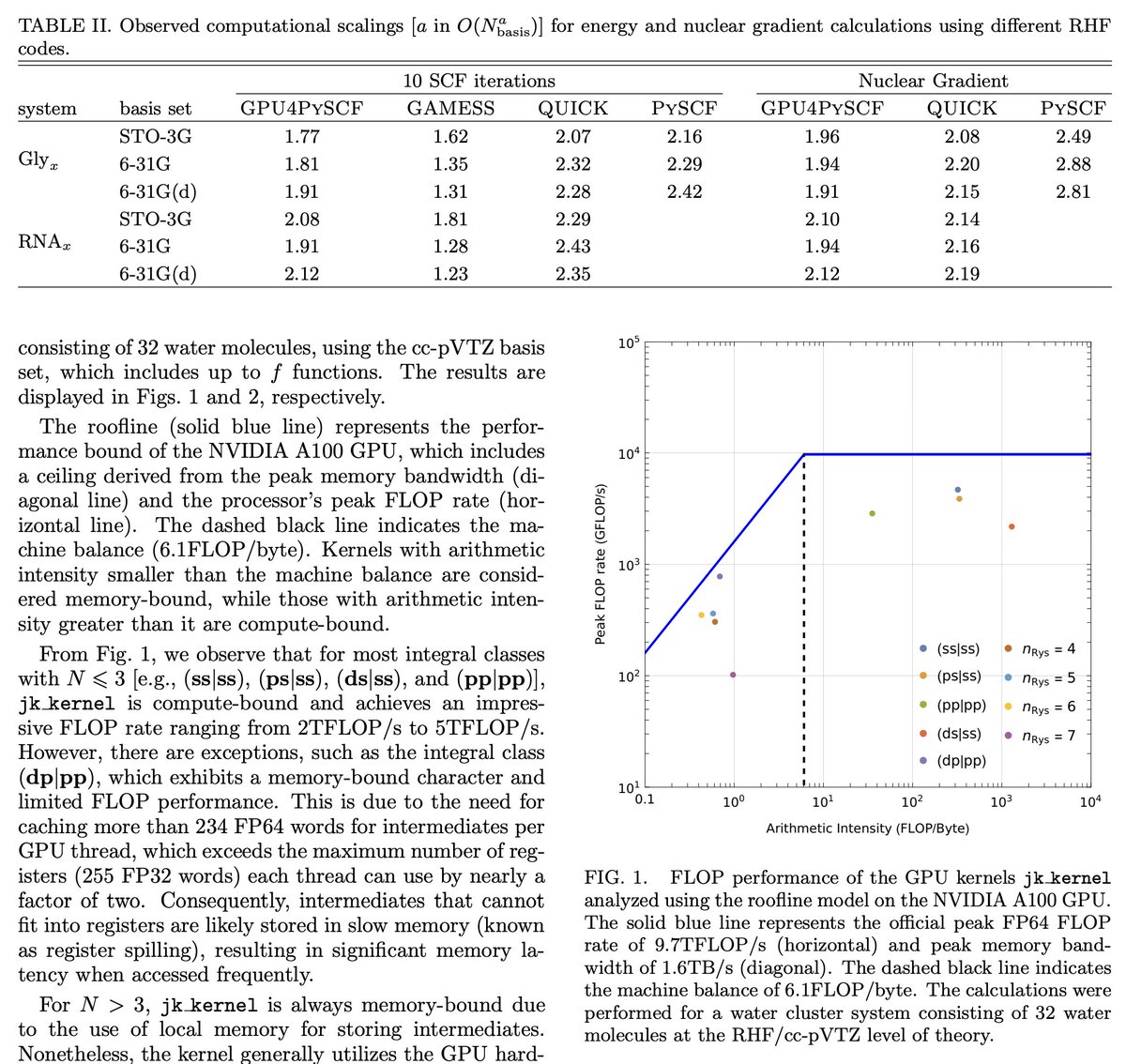
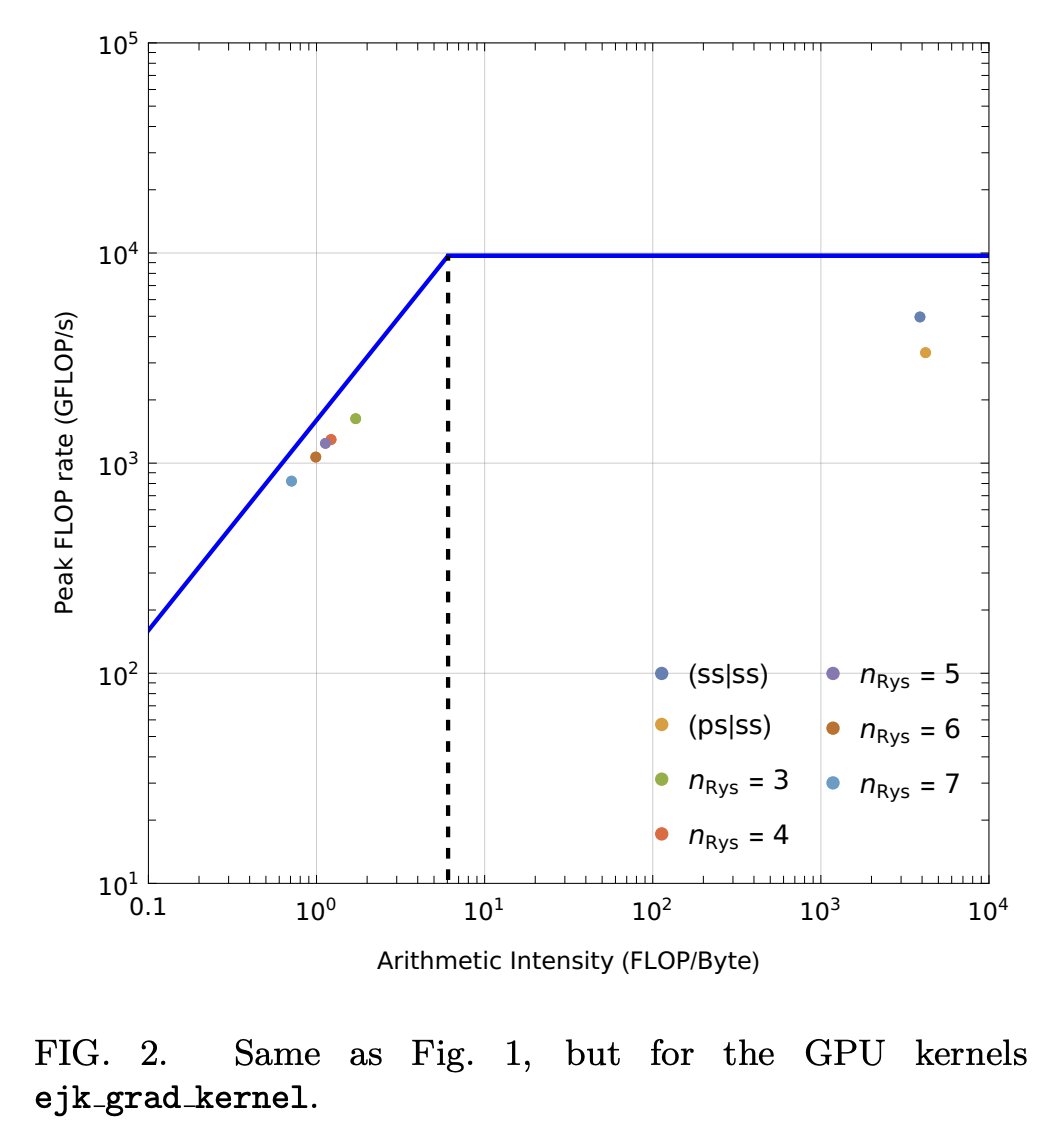
#キャルちゃんのquantphチェック
Pythonベースの化学シミュレーションフレームワーク PySCFに、GPUアクセラレーションを提供するモジュール GPU4PySCFを開発。PySCFのマルチスレッドCPUによるHartree-Fockコードに対し、2桁の大幅な高速化を達成した。
arxiv.org/abs/2407.09700
3
12
1,037

25 Apr 2024
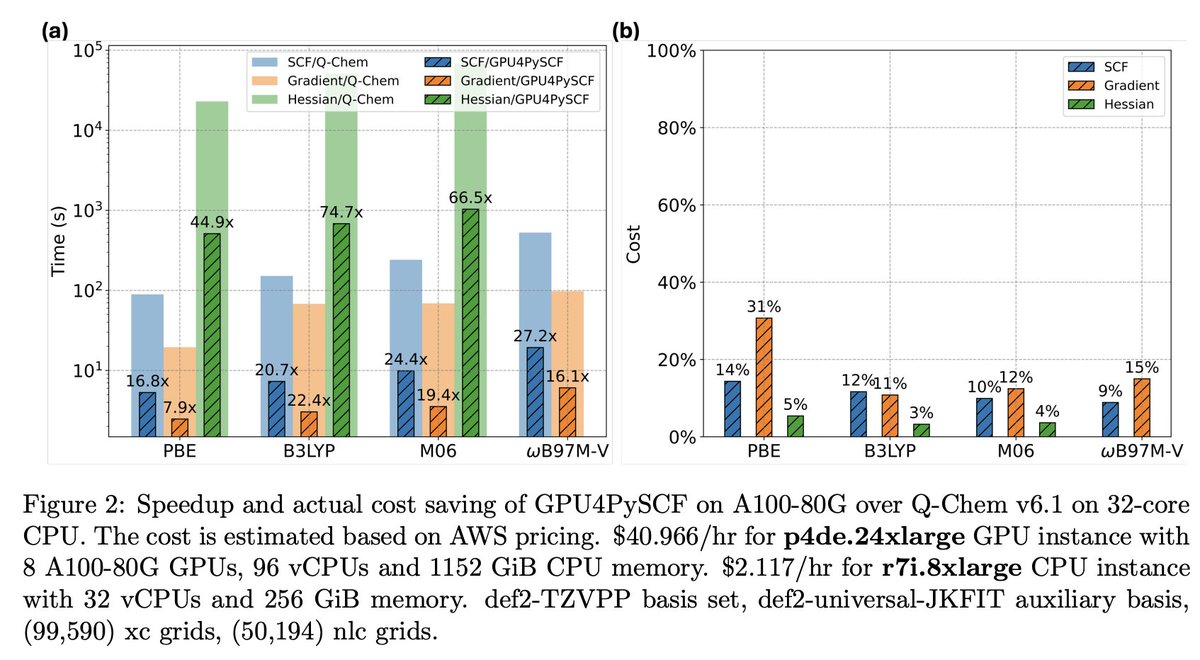
Check out PySCF’s latest preprint in arXiv on the implementation of the GPU acceleration module, GPU4PySCF, which can deliver up to 30 times speedup over a 32-core CPU node, resulting in approximately 90% cost savings for most DFT tasks. 👀
arxiv.org/abs/2404.09452
15
83
7,864
16 Apr 2024
#キャルちゃんのquantphチェック
データ駆動型の化学研究の需要に対応するための、GPUアクセラレーションPythonパッケージ GPU4PySCFを開発。GPUを用いた最新の密度汎函数理論計算を実行した場合、32コアCPUノードと比較して30倍の高速化と、90%のコスト削減を達成した。
arxiv.org/abs/2404.09452
1
13
1,368