Predicting Evolutionary Rate as a Pretraining Task Improves Genome Language Model Representations
1) The paper introduces a novel approach to training genome language models (gLMs) by predicting evolutionary rates as a pretraining task, moving beyond traditional sequence reconstruction methods that often fail to capture meaningful biological signals.
2) Two novel pretraining tasks are proposed: Current Evolution Prediction (CEP), which predicts evolutionary rates autoregressively like next token prediction, and Masked Evolution Modeling (MEM), which predicts rates using bidirectional context like masked language modeling.
3) These evolutionary prediction tasks can be seamlessly composed with existing sequence reconstruction tasks (NTP and MLM), enabling controlled comparisons between training on sequence only, evolution only, or both combined.
4) The authors developed "Gamba" models (ArGamba and Bi-Gamba) ranging from 4M to 66M parameters, demonstrating that relatively small models pretrained on evolutionary rates can compete with much larger existing gLMs like Evo2 (7B parameters) on several benchmarks.
5) A comprehensive suite of biologically grounded zero-shot benchmarks was created to evaluate model representations, including distinguishing functional genomic regions under increasing difficulty settings and classifying ATG codons in different functional contexts.
6) Key finding: Models pretrained on both sequence and evolutionary rate consistently outperform sequence-only models across most tasks, with evolutionary rate-only models also showing strong performance, establishing evolution as a critical training signal for genome-scale models.
7) The work addresses major gaps in gLM evaluation by measuring zero-shot performance without supervised classifiers, including randomly-initialized baselines, and using multiple negative sets of systematically increasing difficulty to reveal what signals models actually learn.
8) On variant effect prediction benchmarks, evolutionary rate prediction provides substantially stronger signal than sequence likelihood alone, with Gamba models matching or outperforming Evo2 on several tasks despite being orders of magnitude smaller.
💻Code: github.com/macaes-lab/gamba
📜Paper: biorxiv.org/content/10.64898…
#Genomics #Bioinformatics #MachineLearning #ComputationalBiology #GenomeLanguageModels #EvolutionaryGenomics #DeepLearning #Pretraining #VariantEffectPrediction
2
38
2,386

Jan 18
A comprehensive survey of genome language models in bioinformatics. #GenomeLanguageModels #gMLs #ModelsReview #Bioinformatics #Genomics #BriefingsInBioinformatics
academic.oup.com/bib/article…
9
57
3,518

19 Sep 2025
Generative design of novel bacteriophages with genome language models
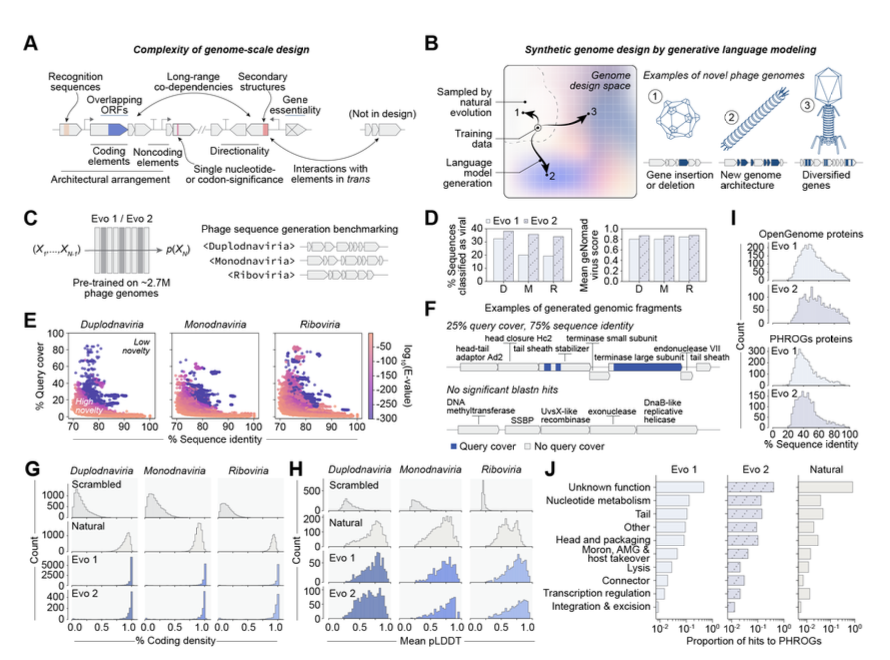
1. A novel study leverages genome language models, specifically Evo 1 and Evo 2, to generate viable bacteriophage genomes, marking the first successful generative design of whole-genome sequences for bacteriophages. This opens new avenues for synthetic biology and phage therapy.
2. The researchers used the lytic phage ΦX174 as a template, applying advanced computational techniques to create phage genomes with significant evolutionary novelty. Experimental validation yielded 16 viable phages, demonstrating the robustness of the generative design approach.
3. Cryo-electron microscopy revealed that one of the generated phages utilizes an evolutionarily distant DNA packaging protein within its capsid, showcasing the potential for designing phages with novel structural features.
4. Multiple generated phages exhibited higher fitness than ΦX174 in growth competitions and faster lysis kinetics. A cocktail of these phages rapidly overcame ΦX174-resistance in three E. coli strains, highlighting the potential for combating antibiotic-resistant bacterial pathogens.
5. The study establishes a generalizable framework for designing synthetic bacteriophages with user-specified constraints, laying a foundation for the generative design of more complex biological systems. This work has broad implications for biotechnology and medicine.
📜Paper: biorxiv.org/content/10.1101/…
#GenomeLanguageModels #Bacteriophages #SyntheticBiology #PhageTherapy #EvolutionaryNovelty
12
49
4,545
25 May 2025
From Likelihood to Fitness: Improving Variant Effect Prediction in Protein and Genome Language Models
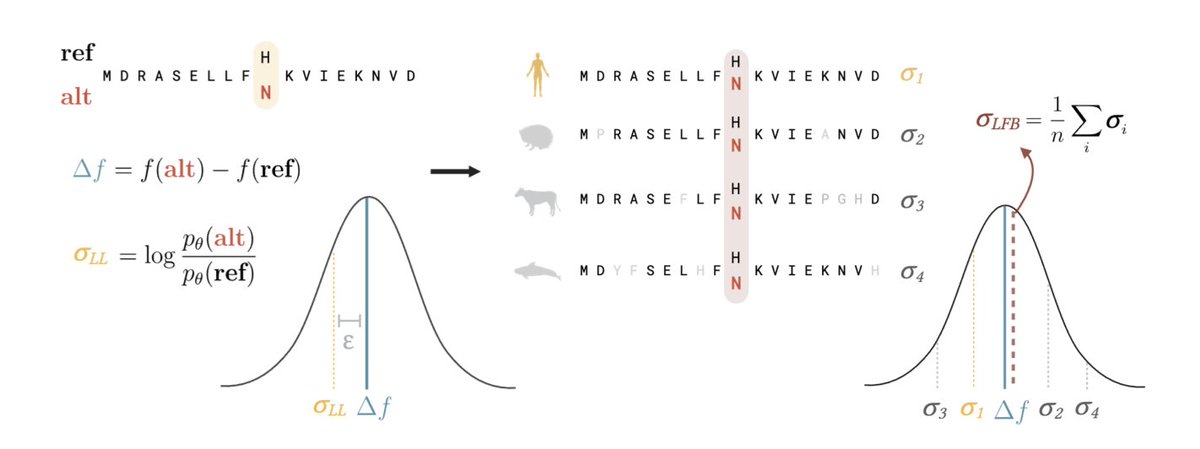
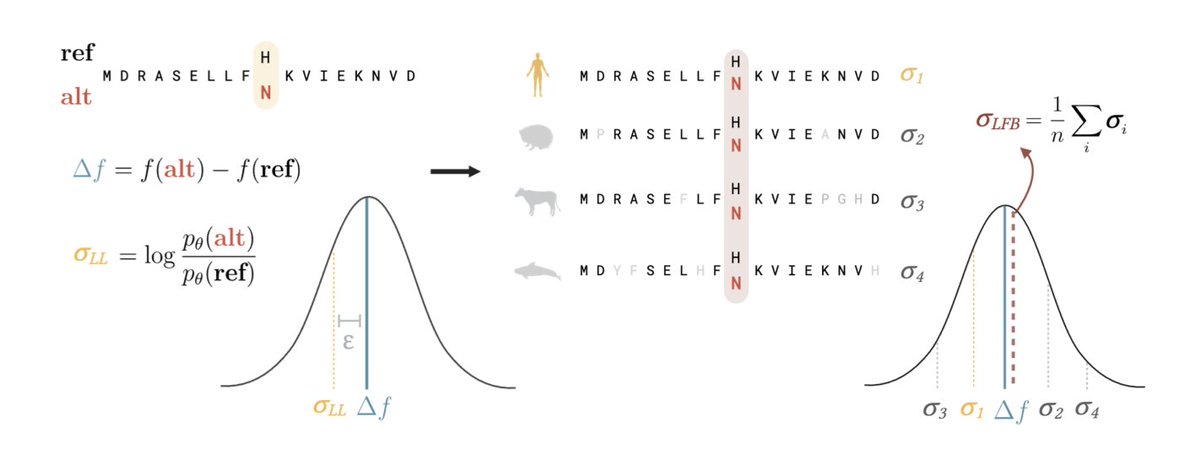
1.This study introduces Likelihood-Fitness Bridging (LFB), a method that improves variant effect prediction in protein and genome language models (pLMs and gLMs) by averaging over phylogenetically related sequences to reduce noise and bias in likelihood-based fitness estimates.
2.LFB does not require retraining or architecture modifications—it operates post hoc on any pretrained generative model, including ESM-2, ProGen2, and Evo 2 families, making it highly practical and scalable.
3.The central insight is that sequence likelihood reflects not only fitness, but also phylogenetic structure and dataset biases. These confounding factors become more pronounced in larger models, which may explain performance plateaus in variant prediction despite improved sequence modeling.
4.Using an Ornstein–Uhlenbeck model of evolution, the authors formalize how averaging log-likelihood changes across homologous sequences suppresses noise introduced by genetic drift, yielding a lower variance estimator of true fitness.
5.When applied to variant classification using clinical labels, LFB consistently improves performance across model families. For ESM-2, LFB elevates AUCs from 0.653 (8M) to 0.889, and from 0.895 (15B) to 0.938, reversing the performance saturation observed at larger model sizes.
6.On the ProteinGym deep mutational scanning (DMS) benchmark, LFB boosts average Spearman correlations across all mutation types. Notably, the 8M ESM-2 model with LFB outperforms the original 35M model, and the 15B model becomes the most accurate overall when LFB is applied.
7.LFB is especially beneficial for larger models that are more susceptible to capturing phylogenetic signals. This confirms that model scale alone doesn't ensure predictive power unless confounding signals are addressed.
8.The method shows broad improvements across mutation types, assays (binding, expression, fitness), and sequence identity thresholds. Surprisingly, strong performance can be retained using as few as 10 homologs per variant.
9.LFB also enhances the separation between pathogenic and benign clinical variants. It reduces the false classification of benign variants as pathogenic, improving the reliability of predictions in a clinical genomics context.
10.While LFB was validated mainly on coding regions and substitution variants, its general framework opens doors for more advanced inference strategies, and points toward principled ways to disentangle fitness from dataset artifacts.
💻Code: github.com/DiasFrazerGroup/l…
📜Paper: biorxiv.org/content/10.1101/…
#VariantEffectPrediction #ProteinLanguageModels #GenomeLanguageModels #LFB #Bioinformatics #DeepLearning #ESM2 #ProGen2 #Evo2 #Genomics #FitnessEstimation #PrecisionMedicine
9
36
2,105
25 May 2025
From Likelihood to Fitness: Improving Variant Effect Prediction in Protein and Genome Language Models
1.This study introduces Likelihood-Fitness Bridging (LFB), a method that improves variant effect prediction in protein and genome language models (pLMs and gLMs) by averaging over phylogenetically related sequences to reduce noise and bias in likelihood-based fitness estimates.
2.LFB does not require retraining or architecture modifications—it operates post hoc on any pretrained generative model, including ESM-2, ProGen2, and Evo 2 families, making it highly practical and scalable.
3.The central insight is that sequence likelihood reflects not only fitness, but also phylogenetic structure and dataset biases. These confounding factors become more pronounced in larger models, which may explain performance plateaus in variant prediction despite improved sequence modeling.
4.Using an Ornstein–Uhlenbeck model of evolution, the authors formalize how averaging log-likelihood changes across homologous sequences suppresses noise introduced by genetic drift, yielding a lower variance estimator of true fitness.
5.When applied to variant classification using clinical labels, LFB consistently improves performance across model families. For ESM-2, LFB elevates AUCs from 0.653 (8M) to 0.889, and from 0.895 (15B) to 0.938, reversing the performance saturation observed at larger model sizes.
6.On the ProteinGym deep mutational scanning (DMS) benchmark, LFB boosts average Spearman correlations across all mutation types. Notably, the 8M ESM-2 model with LFB outperforms the original 35M model, and the 15B model becomes the most accurate overall when LFB is applied.
7.LFB is especially beneficial for larger models that are more susceptible to capturing phylogenetic signals. This confirms that model scale alone doesn't ensure predictive power unless confounding signals are addressed.
8.The method shows broad improvements across mutation types, assays (binding, expression, fitness), and sequence identity thresholds. Surprisingly, strong performance can be retained using as few as 10 homologs per variant.
9.LFB also enhances the separation between pathogenic and benign clinical variants. It reduces the false classification of benign variants as pathogenic, improving the reliability of predictions in a clinical genomics context.
10.While LFB was validated mainly on coding regions and substitution variants, its general framework opens doors for more advanced inference strategies, and points toward principled ways to disentangle fitness from dataset artifacts.
💻Code: github.com/DiasFrazerGroup/l…
📜Paper: biorxiv.org/content/10.1101/…
#VariantEffectPrediction #ProteinLanguageModels #GenomeLanguageModels #LFB #Bioinformatics #DeepLearning #ESM2 #ProGen2 #Evo2 #Genomics #FitnessEstimation #PrecisionMedicine
2
17
88
21,894
15 Mar 2025
Transformers and genome language models @NatMachIntell
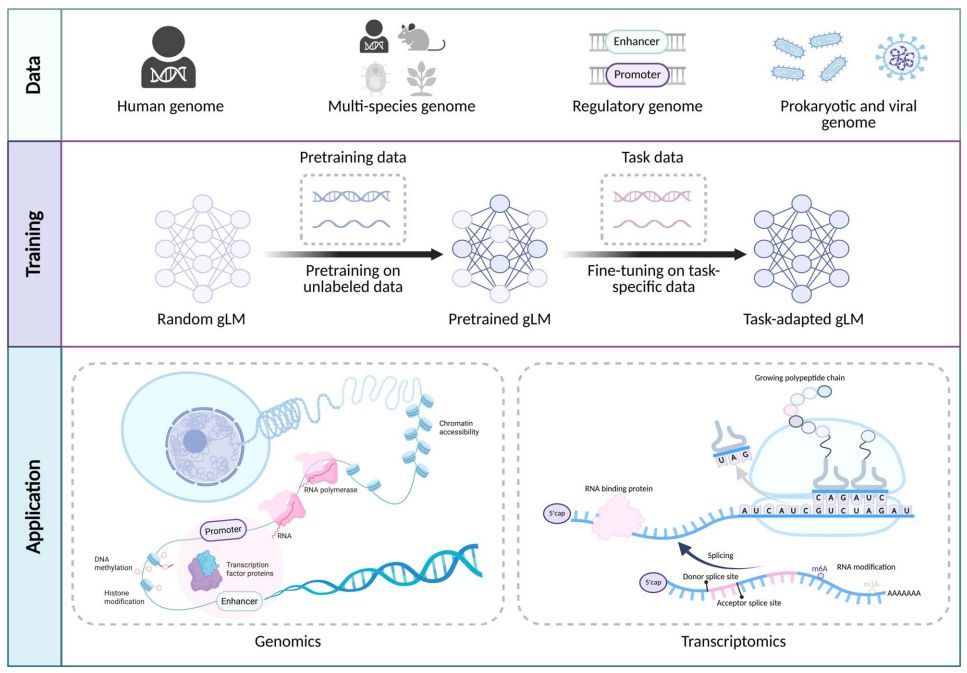
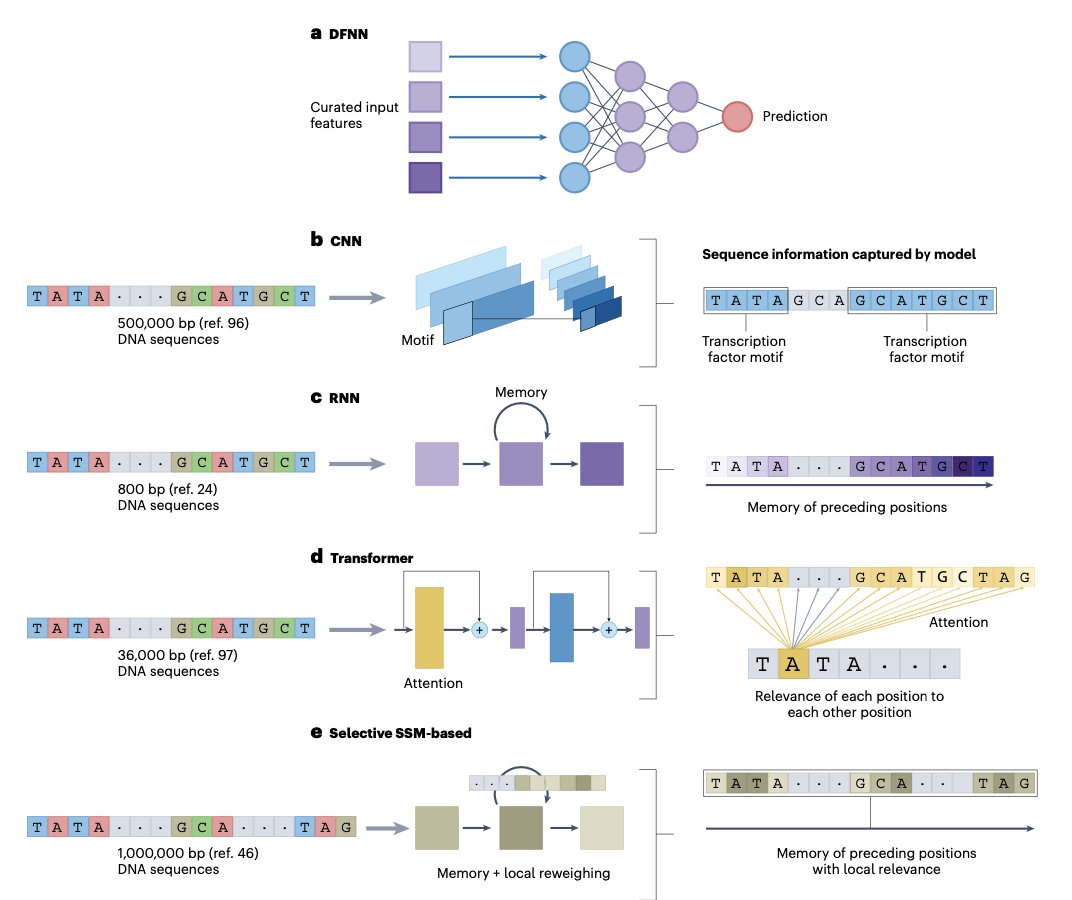
- Transformer-based language models are transforming genomics, just as they did for natural language processing. This review explores the rise of genome language models (gLMs) and their ability to extract meaningful patterns from DNA sequences using deep learning.
- gLMs leverage self-supervised pretraining to learn genomic features without labeled data, enabling zero-shot and few-shot learning. This makes them particularly valuable for studying complex genomic regulatory mechanisms.
- Unlike traditional convolutional neural networks (CNNs) and recurrent neural networks (RNNs), transformers excel in modeling long-range dependencies in DNA, capturing interactions across distant genomic regions that influence gene expression.
- The review highlights key applications of gLMs, including functional genomics, variant effect prediction, gene expression modeling, and chromatin accessibility analysis. These models offer a promising approach to deciphering the "regulatory grammar" of the genome.
- Tokenization is a critical aspect of genomic modeling. The review compares one-hot encoding, k-mer tokenization, and byte-pair encoding (BPE), each with trade-offs in capturing genomic structure.
- While transformers have dominated recent advances, the authors also discuss alternative architectures such as state space models (SSMs), which may provide computational advantages for large-scale genomic datasets.
- The future of gLMs lies in integrating multimodal data, expanding model interpretability, and improving efficiency. As genomic datasets grow, scaling up these models will be key to unlocking new biological insights.
📜Paper: nature.com/articles/s42256-0…
#Genomics #AI #DeepLearning #Transformers #GenomeLanguageModels #ComputationalBiology #Bioinformatics
12
66
5,645
15 Mar 2025
Transformers and genome language models @NatMachIntell
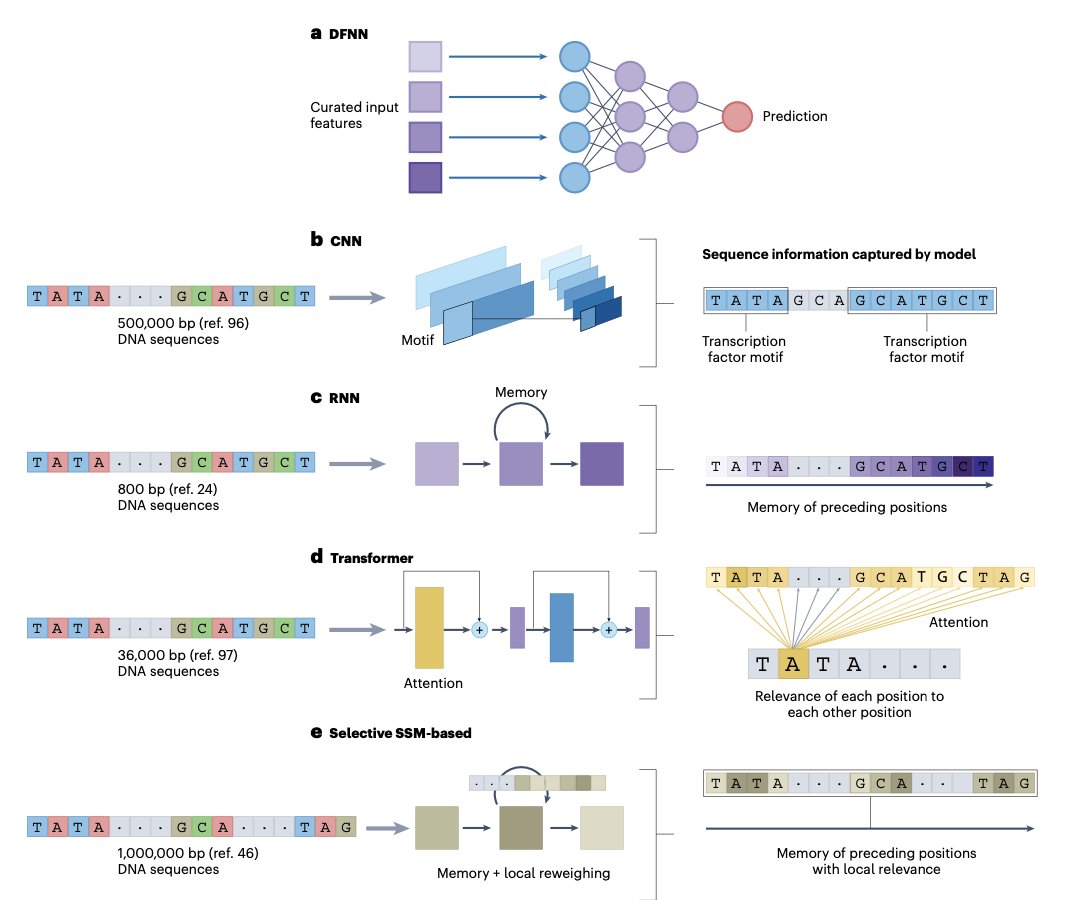
- Transformer-based language models are transforming genomics, just as they did for natural language processing. This review explores the rise of genome language models (gLMs) and their ability to extract meaningful patterns from DNA sequences using deep learning.
- gLMs leverage self-supervised pretraining to learn genomic features without labeled data, enabling zero-shot and few-shot learning. This makes them particularly valuable for studying complex genomic regulatory mechanisms.
- Unlike traditional convolutional neural networks (CNNs) and recurrent neural networks (RNNs), transformers excel in modeling long-range dependencies in DNA, capturing interactions across distant genomic regions that influence gene expression.
- The review highlights key applications of gLMs, including functional genomics, variant effect prediction, gene expression modeling, and chromatin accessibility analysis. These models offer a promising approach to deciphering the "regulatory grammar" of the genome.
- Tokenization is a critical aspect of genomic modeling. The review compares one-hot encoding, k-mer tokenization, and byte-pair encoding (BPE), each with trade-offs in capturing genomic structure.
- While transformers have dominated recent advances, the authors also discuss alternative architectures such as state space models (SSMs), which may provide computational advantages for large-scale genomic datasets.
- The future of gLMs lies in integrating multimodal data, expanding model interpretability, and improving efficiency. As genomic datasets grow, scaling up these models will be key to unlocking new biological insights.
📜Paper: nature.com/articles/s42256-0…
#Genomics #AI #DeepLearning #Transformers #GenomeLanguageModels #ComputationalBiology #Bioinformatics
3
938