Towards a Generative Protein Evolution Machine with DPLM-Evo
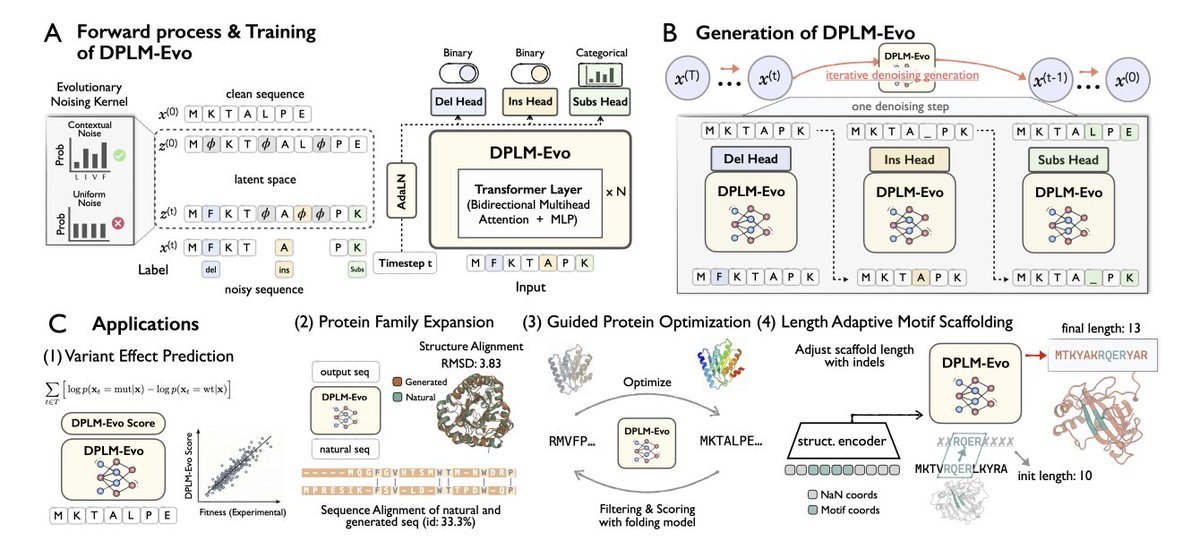
1. DPLM-Evo reframes protein diffusion generation around explicit evolutionary edits: substitutions, insertions, and deletions (indels). This addresses a mismatch in prior DPLMs where proteins “emerge from masks,” despite real evolution proceeding via accumulated edits.
2. Core idea: decouple a fixed-size latent alignment space from the variable-length observed sequence. Indels become gap ↔ residue transitions in the latent space, making variable-length diffusion tractable while keeping compute close to fixed-length models.
3. Architecture: the model denoises in observed sequence space but predicts three edit signals per position via separate heads: (i) amino-acid distribution for substitution, (ii) deletion probability, and (iii) insertion probability (insert to the right; residue identity comes from the substitution head).
4. A key innovation is the contextualized evolutionary noising kernel for substitutions. Instead of uniform random corruption, substitutions are corrupted using a context-dependent distribution derived from the model’s own predictions (after a warmup), producing more biologically plausible mutation patterns during training.
5. This contextualized corruption materially matters: an ablation replacing it with uniform corruption drops ProteinGym average Spearman from 0.42 to 0.295; a static BLOSUM-based kernel lands in-between (~0.35), supporting the claim that context-aware mutation noise better matches evolutionary constraints.
6. Understanding task highlight: DPLM-Evo achieves state-of-the-art mutation effect prediction on ProteinGym among single-sequence foundation models (217 DMS assays). Scoring is “substitution-native”: it directly reads substitution probabilities at mutated sites without masking them, avoiding an artificial mask-token scoring mismatch.
7. Indel effect prediction: on the ProteinGym indel benchmark, DPLM-Evo reaches 0.495 Spearman, outperforming strong single-sequence baselines (e.g., ProGen2 M 0.464) and approaching MSA-based methods (PoET 0.517, ProFam ensemble 0.530), suggesting explicit indel modeling transfers to indel fitness estimation.
8. Generation: DPLM-Evo enables variable-length unconditional protein generation via evolutionary denoising (sub/ins/del), starting from a learned prior rather than an all-mask state. It maintains strong foldability (ESMFold pLDDT ~83.6, comparable to DPLM) while improving diversity and reducing repetition/mode collapse.
9. Conditional design: in motif scaffolding, DPLM-Evo can dynamically adjust scaffold length during sampling (via insertion/deletion heads) while keeping motif residues fixed, avoiding manual enumeration of scaffold lengths required by fixed-length generators; it improves solved motif counts and success rate in zero-shot and further with continued finetuning.
10. Edit-trajectory applications: the model supports post-editing and optimization as explicit evolutionary trajectories. Case studies include in-silico “family expansion” (large sequence divergence while preserving fold) and directed evolution of GFP, where enabling indels improved structural scores faster and higher than substitution-only and an ESM-2 baseline under the same search/filtering protocol.
📜Paper: arxiv.org/abs/2605.00182
#ComputationalBiology #ProteinDesign #ProteinLanguageModels #DiffusionModels #GenerativeAI #MachineLearning #Bioinformatics #DirectedEvolution #ProteinEngineering #VariantEffectPrediction
1
6
26
2,415
eSIG-Net: An interaction language model that decodes the protein code of single mutations @naturemethods
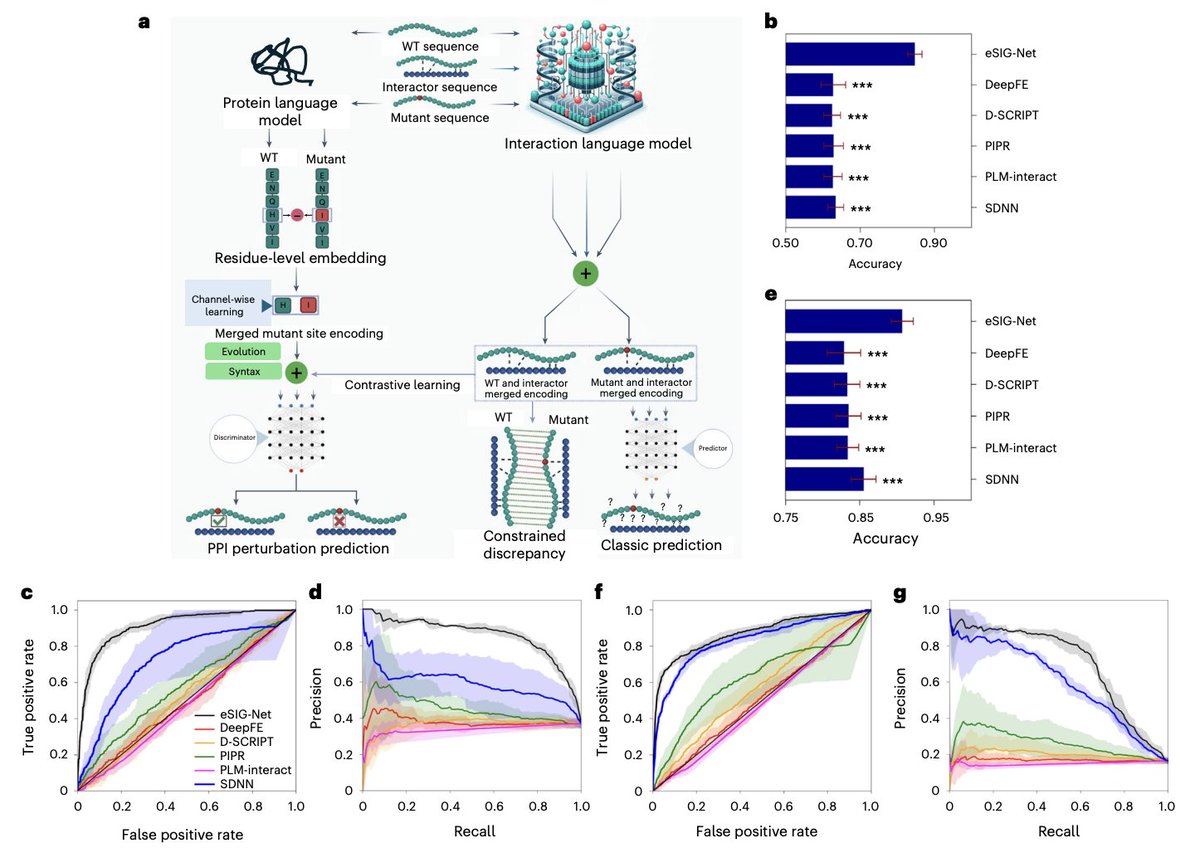
1. eSIG-Net is a mutation-centric interaction language model designed to predict whether a single missense mutation rewires a specific protein–protein interaction (PPI), using sequence information alone and explicitly modeling the subtle wild-type (WT) vs mutant discrepancy.
2. The paper frames single-mutation effects on PPIs as an “interaction cliff” problem: tiny sequence edits can cause large, hard-to-predict interaction changes, which standard sequence/structure predictors often miss because they do not directly learn WT–mutant contrasts.
3. Core design: eSIG-Net operates on triplets (WT protein, mutant protein, interactor) and jointly encodes (WT, interactor) and (mutant, interactor), then learns a constrained discrepancy between the two merged representations to highlight mutation-driven interaction shifts.
4. Key innovation module I: constrained discrepancy learning. It adds a loss that encourages the model to learn different embedding distances depending on whether the mutation is truly perturbing (change) or non-perturbing (no change), improving separation when training data is limited.
5. Key innovation module II: syntax-aware and evolution-aware mutation-site encoding from a protein language model (ESM2). Instead of pooling whole-sequence embeddings, it extracts residue-level embeddings at the mutation site and learns a focused representation of the WT→mutant change via channel-wise learning.
6. Final prediction is produced by a discriminator that integrates: (a) WT–interactor merged encoding, (b) mutant–interactor merged encoding, and (c) mutation-site encoding—aiming to predict interaction perturbation directly (not just PPI existence).
7. Benchmarking on a disease mutation PPI dataset (1,633 triplets; 606 perturbed and 1,027 non-perturbed PPIs): eSIG-Net reports accuracy 0.85 ± 0.02, outperforming sequence-based PPI methods (best baseline accuracy 0.63 ± 0.02), with ROC-AUC 0.91 ± 0.02 and average precision 0.86 ± 0.01.
8. Robustness on an imbalanced population variant dataset from gnomAD/ExAC (1,650 variants; ~16% positives; 4,020 PPIs): eSIG-Net maintains strong performance (reported accuracy up to 0.90 ± 0.01), supporting generalization beyond disease-enriched mutations.
9. Structure-based comparisons: AlphaFold-derived and other mutation-centric structure tools (FoldDock, MutaBind2, BeAtMuSiC, GeoPPI, TopNetTree, PIONEER) reach ~≤60% accuracy on the disease dataset, while eSIG-Net remains substantially higher; the paper highlights that WT vs mutant complex structures can be globally near-indistinguishable, limiting structure-only signals for edgetic effects.
10. Biological interpretability/use cases: eSIG-Net can propose mutation-specific, partner-specific “edgetic” mechanisms (example: pleiotropic TPM3 mutations predicted to differentially perturb interaction with HSF2), and it supports large-scale in silico mutagenesis for variant-of-unknown-significance prioritization; the paper also presents cohort-level associations where predicted PPI gene pairs relate to survival and immunotherapy response contexts.
💻Code: github.com/Stephen-Yi-Labora…
📜Paper: doi.org/10.1038/s41592-026-0…
#ComputationalBiology #ProteinInteractions #PPI #VariantEffectPrediction #ProteinLanguageModels #DeepLearning #Genomics #MissenseVariants #Interactome #NetworkBiology
3
16
1,547
Proteome-wide prediction of the functional impact of missense variants with ProteoCast @NatureComms
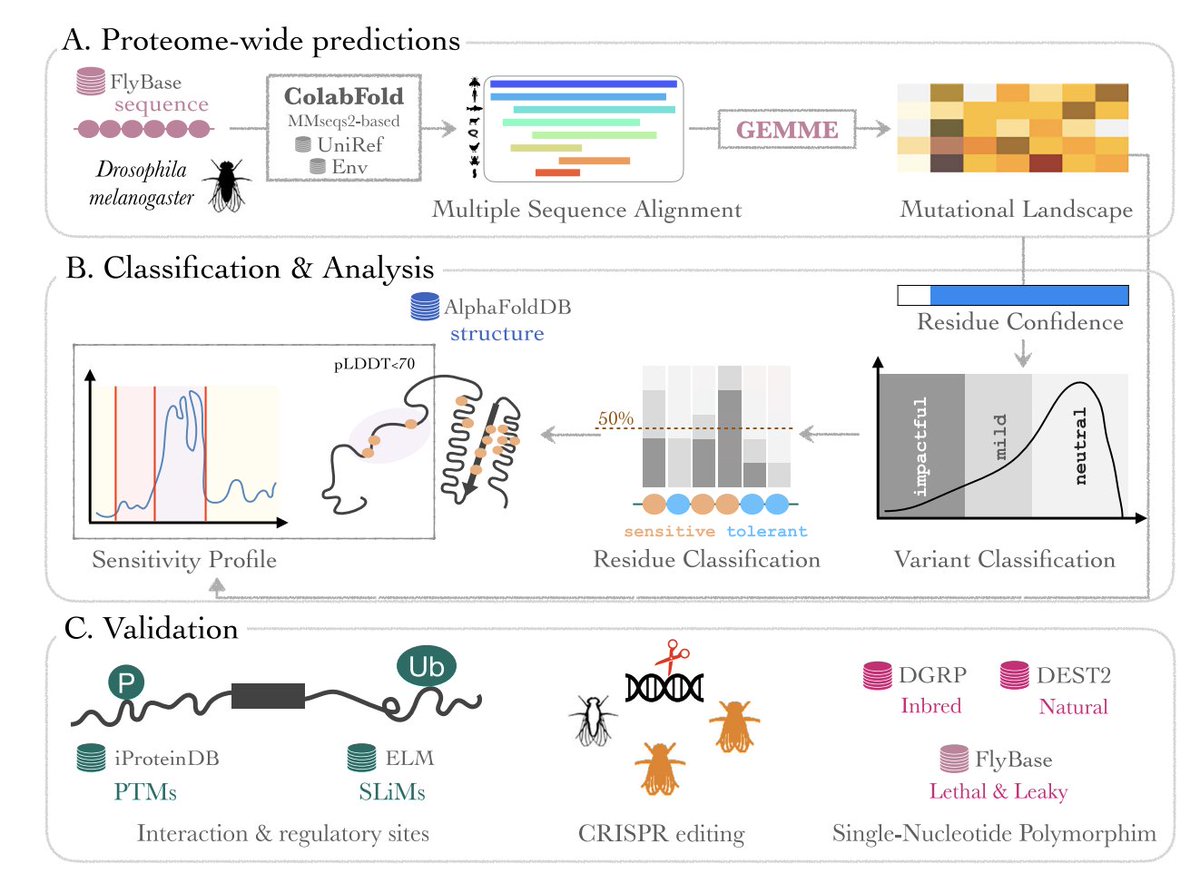
1. ProteoCast is an automated, scalable framework that predicts missense variant impact across entire proteomes using evolutionary information alone, emphasizing interpretability and practical prioritization for genome editing.
2. The study applies ProteoCast to 22,169 Drosophila melanogaster proteoforms (including isoforms), generating predicted effects for ~293 million possible missense substitutions and serving them through an interactive database.
3. Core methodological idea: start from ColabFold-generated MSAs, run GEMME to obtain full single-site mutational landscapes, then add (a) reliability/confidence metrics, (b) protein-specific variant classification, and (c) residue/segment-level functional site discovery.
4. A key innovation is adaptive per-protein variant classification. ProteoCast fits a 3-component Gaussian mixture model to each protein’s raw score distribution and classifies variants into neutral, mild, or impactful using protein-specific thresholds (rather than a universal cutoff).
5. Reliability is handled explicitly: proteins with <200 homologs are flagged; residues are flagged as unreliable using a combined criterion (score dispersion across 19 substitutions, conservation, alignment coverage, and fraction of substitutions observed), then smoothed along sequence to reduce cutoff artifacts.
6. Large-scale validation in fly: benchmarked against ~386k variants spanning natural polymorphisms (DGRP inbred lines; DEST2 wild populations) and FlyBase EMS mutations with phenotypes. ProteoCast classifies 85% of known lethal mutations as impactful/mild, while labeling most population variants as neutral (88% DGRP; 82% DEST2), aligning evolutionary constraint with organismal fitness.
7. Experimental confirmation via CRISPR editing: ProteoCast-guided edits in the essential NAD-biosynthesis enzyme Naprt produced the predicted outcomes—three “impactful” substitutions could not be homozygosed (consistent with recessive lethality), while two “neutral” substitutions yielded viable homozygotes, including challenging physicochemical changes.
8. Beyond variant scoring, ProteoCast uses mutational sensitivity (mean effect across substitutions) mapped onto AlphaFold models to highlight constrained 3D regions; it also shows that low AlphaFold pLDDT does not imply lack of evolutionary constraint (many low-pLDDT residues still have confident variant predictions).
9. A second major innovation is functional site discovery in disordered regions: ProteoCast segments the 1D mutational-sensitivity profile with changepoint detection (FPOP) to find locally elevated sensitivity “islands,” which enrich for regulatory/binding features. Proteome-wide, 63% of PTM sites and strong fractions of SLiMs overlap elevated-sensitivity segments (85% partial overlap; 61% full inclusion).
10. Generalization to human/benchmarks: on ClinVar (~63k variants), ProteoCast provides predictions for ~99% of variants and achieves a balanced sensitivity/specificity profile; compared with EVE, it offers higher coverage and avoids discarding large fractions as “uncertain,” while remaining competitive in discrimination. It also performs strongly for disordered binding-site detection (CAID3 Binding-IDR) using segmentation-based signals.
💻Code: hub.docker.com/r/marinaabaka…
📜Paper: doi.org/10.1038/s41467-026-7…
#ComputationalBiology #Bioinformatics #VariantEffectPrediction #ProteinEvolution #MSA #Drosophila #CRISPR #AlphaFold #FunctionalGenomics #MissenseVariants
4
24
1,973
MIMIC: A Generative Multimodal Foundation Model for Biomolecules
1. The paper introduces MIMIC, a 1B-parameter generative multimodal foundation model that learns a joint distribution over biomolecular “state” across the central dogma—genome, transcriptome, and proteome—so it can infer missing modalities and support both prediction and constrained design.
2. A key enabling contribution is LORE, a newly curated aligned dataset with partially observed multimodal snapshots: ~15.5M proteins and ~13M RNA transcripts from >6000 organisms, plus >4B tokens of biomedical/functional/context text. LORE aligns signals into consistent coordinate systems (nucleotide- and residue-aligned tracks) rather than siloed corpora.
3. MIMIC’s core architectural idea is a split-track encoder-decoder Transformer: nucleic-acid and amino-acid tracks each sum multiple co-aligned per-position modalities (e.g., sequence phyloP structural labels) into a single track representation, keeping length manageable even as modalities grow.
4. It uses RoPE with “group-reset” positional indexing (positions restart per track group) so attention reflects within-track distances, while unaligned global information (semantic/context text) is placed in separate token groups. Learnable register tokens are trained to capture compressed global state via reconstruction under token dropout.
5. The asymmetric encoder/decoder design is central: the encoder can take long context (trained up to 10,000 tokens via curriculum), while the decoder has a smaller context window (~1,000 tokens) and can reconstruct/generate arbitrary target modalities without task-specific heads—enabling “condition on any subset, generate any subset.”
6. On multimodal sequence completion, conditioning on auxiliary modalities consistently improves reconstruction. For proteins, conditioning on structure/surface yields top inpainting accuracy vs protein sequence-only LMs (ProtBERT, ESM-2, ESM-C, ESM3-open). For transcripts, conditioning on aligned genomic modalities outperforms long-context genome FMs (NTv3, Evo2) for masked nucleotide recovery in both introns and exons.
7. For representation transfer, encoder embeddings with simple supervised probes reach state-of-the-art results across PFMBench (protein function/structure/interaction/developability) and mRNABench (mRNA function/localization/translation/variant effect). Gains are especially notable in tasks plausibly helped by protein-aligned or surface-chemistry supervision (e.g., ligand binding, developability).
8. The generative setup enables an interpretable variant-effect approach: instead of only probing embeddings, they use MIMIC-generated wild-type phyloP and mutant–wild-type predicted phyloP differences as a 2D feature space for logistic regression, substantially improving variant effect prediction—especially on complex variants—while remaining low-dimensional and interpretable.
9. MIMIC achieves state-of-the-art splicing prediction and shows a practical advantage of “isoform-aware” inference: conditioning on transcript boundaries (TSS/TES) improves transcript-level splice structure recovery and reduces false-positive splice junctions, highlighting that splicing is better modeled as transcript architecture rather than independent per-site labels.
10. The same conditional generation supports constrained design. For RNA, MIMIC proposes compensatory edits that reduce cryptic exon inclusion for the classic HBB IVS-II-654 C>T splice mutation without reverting the causal base, using multimodal conditioning (wild-type splice pattern and optionally wild-type phyloP). For proteins, conditioning jointly on backbone geometry and MaSIF surface fingerprints at binding sites generates diverse sequences with strong in silico support for folding (AlphaFold2 pLDDT) and, in some cases, binding (AlphaFold3 iPTM) for PD-L1 and hACE2 interface design.
11. A distinctive multimodal angle is treating experimental context as a first-class conditioning variable via natural language. On RNA chemical probing (RASP2), context-conditioned generation improves reactivity prediction vs sequence-only and captures differences between in vivo vs in vitro and between probes (e.g., icSHAPE vs DMS). Predicted reactivities can then guide ViennaRNA folding to improve RNA 2D structure accuracy across a test set.
📜Paper: arxiv.org/abs/2604.24506
#ComputationalBiology #Bioinformatics #FoundationModels #MultimodalAI #Genomics #Transcriptomics #Proteomics #ProteinDesign #RNA #Splicing #VariantEffectPrediction

3
34
2,032
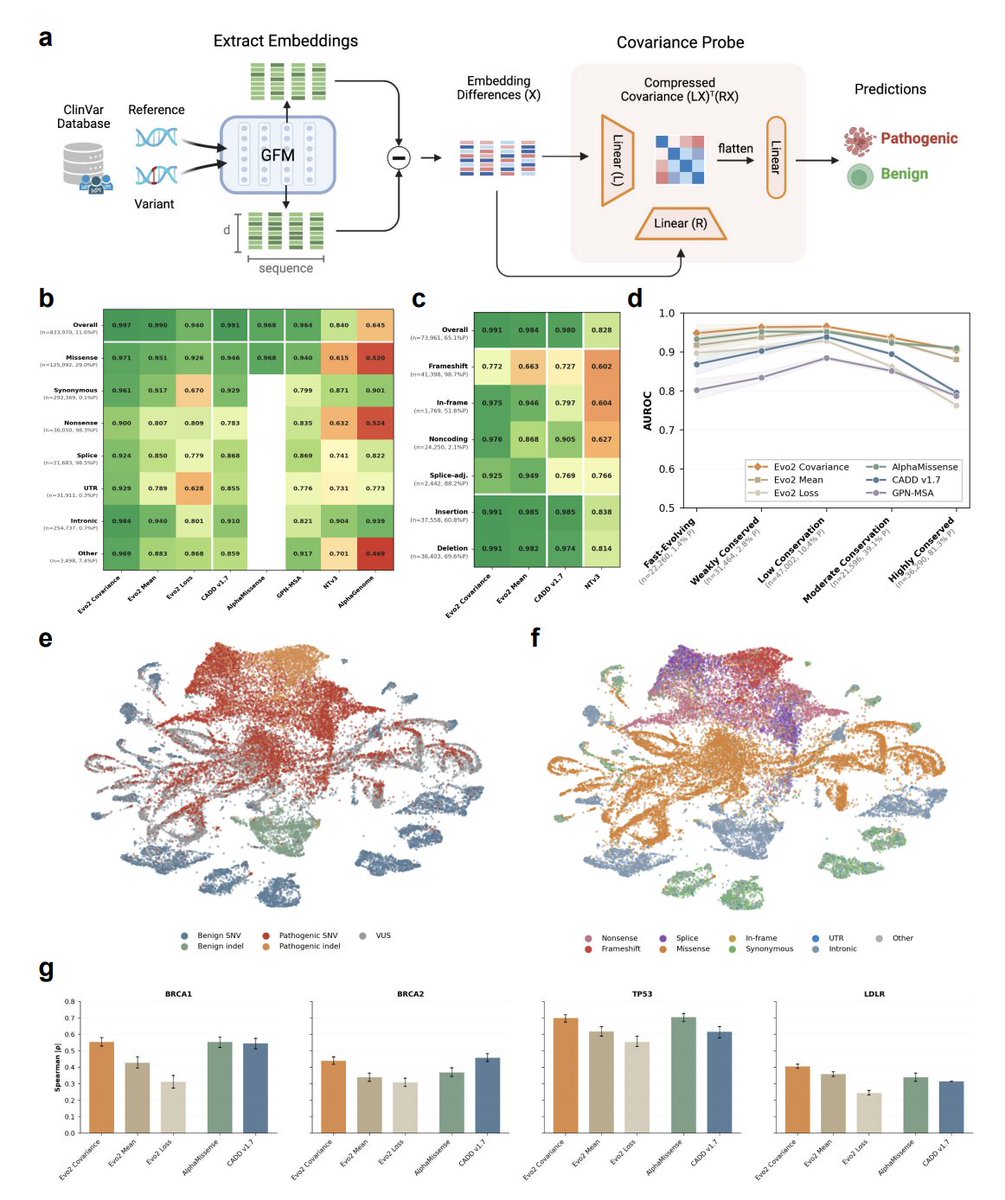
EVEE: Interpretable variant effect prediction from genomic foundation model embeddings
1. EVEE introduces a single embedding-based framework that predicts variant pathogenicity across essentially all common consequence types (coding and non-coding SNVs, plus indels) while also producing structured, human-readable mechanistic rationales.
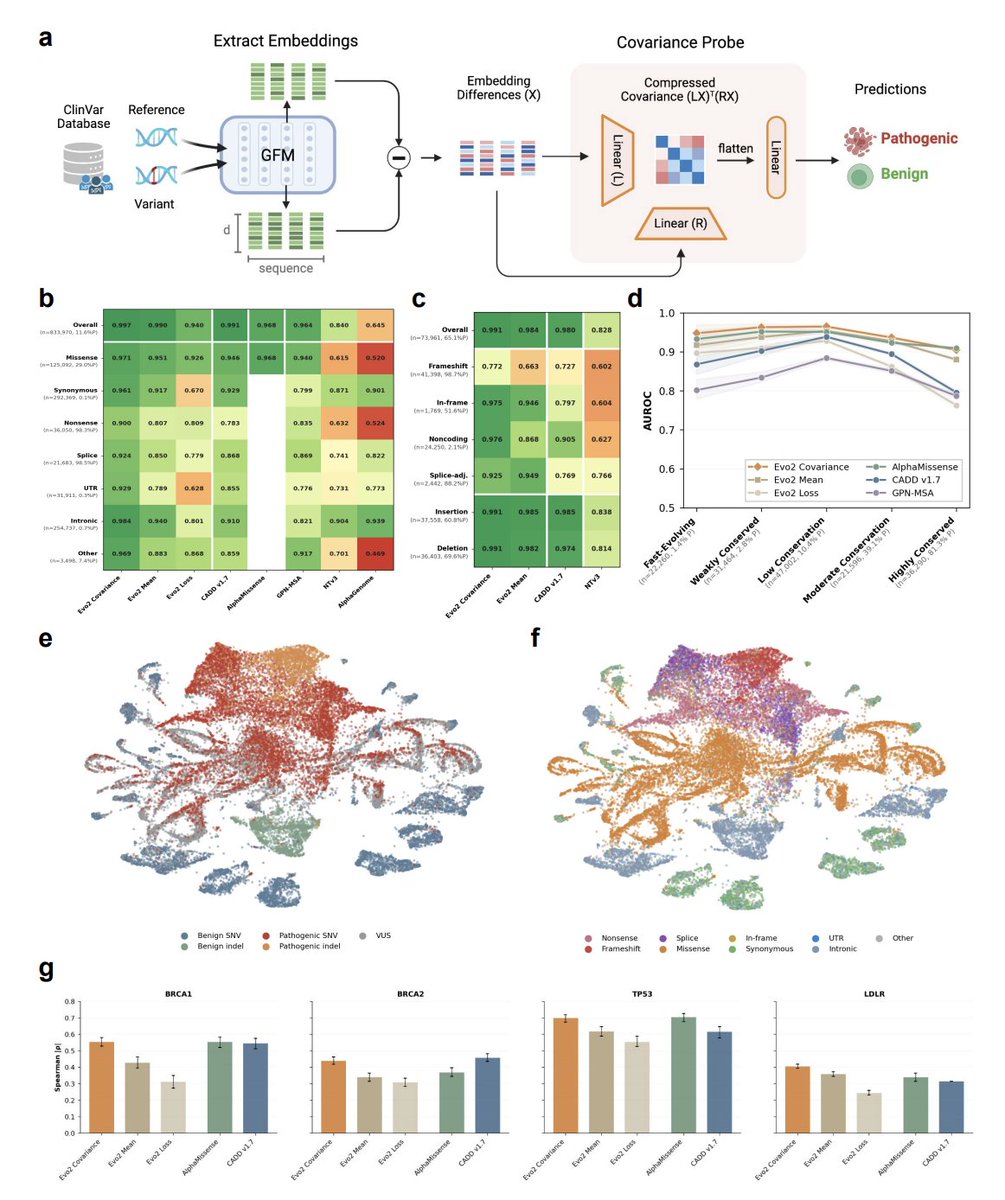
2. The core modeling idea is a covariance (Gram-matrix) “probe” on Evo 2 (7B) embeddings: compute per-position embedding deltas (ALT minus REF), then pool with second-order statistics (XᵀX) rather than mean pooling, capturing interactions between embedding dimensions that are lost in standard pooled representations.
3. On 833,970 ClinVar SNVs (genes ≤100 kb; ≥1-star review; gene-level holdout), the Evo 2 covariance probe reports 0.997 overall AUROC, with strong stratified AUROCs across consequence types: missense 0.971, synonymous 0.961, nonsense 0.900, splice 0.924, UTR 0.929, intronic 0.984, other non-coding 0.969—explicitly addressing the “consequence-type prior” confound by evaluating within each type.
4. Despite being trained only on SNVs, the same probe transfers zero-shot to 73,961 ClinVar indels with 0.986 overall AUROC (1 bp 0.989; 2–5 bp 0.991; 6–20 bp 0.984; >20 bp 0.916; insertions 0.991; deletions 0.988). This suggests the learned embedding geometry captures general sequence-disruption principles beyond point substitutions.
5. Robustness analysis shows performance remains high across conservation tiers (phyloP100way), where annotation-heavy and conservation-driven approaches degrade at conservation extremes—supporting the claim that Evo 2 encodes constraint signals that are complementary to classical conservation.
6. Biological generalization is tested via deep mutational scanning transfer on held-out genes (BRCA1, BRCA2, TP53, LDLR). ClinVar-trained embedding probes outperform Evo 2 loss-based scoring, and covariance pooling consistently improves over mean pooling; results are competitive with protein-focused methods, with particularly strong correlation on TP53 (|ρ| ≈ 0.70).
7. Interpretability is built via supervised “annotation probes” trained to decode 251 biological properties from Evo 2 reference embeddings (357 annotation types reported; median AUROC 0.919). For a given variant, EVEE computes an annotation disruption profile: per-annotation deltas between ALT and REF predictions, including effects at the variant position and up to five flanking positions to capture local propagation (e.g., splice boundary disruptions).
8. EVEE then converts the top disruption signals into natural-language explanations using a reasoning LLM, combining variant metadata (gene, consequence, HGVS) with disruption features (e.g., splice acceptor loss, branch-point collapse, polypyrimidine tract weakening). A highlighted example is BRCA1 c.4987−3C>G, where coordinated predicted splice-acceptor machinery disruption aligns with known RNA evidence for aberrant splicing.
9. Explanation quality is evaluated against expert-panel ClinVar text using an LLM-as-judge on 154 ≥3-star variants. Adding Evo 2 probe-derived disruption signals yields the largest incremental gain in judged quality, reaching a composite 3.89/5 (mechanism coverage 3.15; biological accuracy 4.27; specificity 4.15) with the strongest interpreter model tier reported.
10. The resource component matters: EVEE provides precomputed predictions and disruption profiles for 4.2 million ClinVar variants, plus on-demand explanations through an interactive explorer—positioning foundation-model embeddings as a shared substrate for both prediction and mechanistic inspection rather than a score-only black box.
💻Code: github.com/goodfire-ai/evee-…
📜Paper: biorxiv.org/content/10.64898…
#ComputationalGenomics #VariantEffectPrediction #GenomicFoundationModels #ClinVar #Interpretability #DeepLearning #Bioinformatics #GenomicMedicine #Splicing #Indels
1
6
1,022
EVEE: Interpretable variant effect prediction from genomic foundation model embeddings
1. EVEE introduces a single embedding-based framework that predicts variant pathogenicity across essentially all common consequence types (coding and non-coding SNVs, plus indels) while also producing structured, human-readable mechanistic rationales.
2. The core modeling idea is a covariance (Gram-matrix) “probe” on Evo 2 (7B) embeddings: compute per-position embedding deltas (ALT minus REF), then pool with second-order statistics (XᵀX) rather than mean pooling, capturing interactions between embedding dimensions that are lost in standard pooled representations.
3. On 833,970 ClinVar SNVs (genes ≤100 kb; ≥1-star review; gene-level holdout), the Evo 2 covariance probe reports 0.997 overall AUROC, with strong stratified AUROCs across consequence types: missense 0.971, synonymous 0.961, nonsense 0.900, splice 0.924, UTR 0.929, intronic 0.984, other non-coding 0.969—explicitly addressing the “consequence-type prior” confound by evaluating within each type.
4. Despite being trained only on SNVs, the same probe transfers zero-shot to 73,961 ClinVar indels with 0.986 overall AUROC (1 bp 0.989; 2–5 bp 0.991; 6–20 bp 0.984; >20 bp 0.916; insertions 0.991; deletions 0.988). This suggests the learned embedding geometry captures general sequence-disruption principles beyond point substitutions.
5. Robustness analysis shows performance remains high across conservation tiers (phyloP100way), where annotation-heavy and conservation-driven approaches degrade at conservation extremes—supporting the claim that Evo 2 encodes constraint signals that are complementary to classical conservation.
6. Biological generalization is tested via deep mutational scanning transfer on held-out genes (BRCA1, BRCA2, TP53, LDLR). ClinVar-trained embedding probes outperform Evo 2 loss-based scoring, and covariance pooling consistently improves over mean pooling; results are competitive with protein-focused methods, with particularly strong correlation on TP53 (|ρ| ≈ 0.70).
7. Interpretability is built via supervised “annotation probes” trained to decode 251 biological properties from Evo 2 reference embeddings (357 annotation types reported; median AUROC 0.919). For a given variant, EVEE computes an annotation disruption profile: per-annotation deltas between ALT and REF predictions, including effects at the variant position and up to five flanking positions to capture local propagation (e.g., splice boundary disruptions).
8. EVEE then converts the top disruption signals into natural-language explanations using a reasoning LLM, combining variant metadata (gene, consequence, HGVS) with disruption features (e.g., splice acceptor loss, branch-point collapse, polypyrimidine tract weakening). A highlighted example is BRCA1 c.4987−3C>G, where coordinated predicted splice-acceptor machinery disruption aligns with known RNA evidence for aberrant splicing.
9. Explanation quality is evaluated against expert-panel ClinVar text using an LLM-as-judge on 154 ≥3-star variants. Adding Evo 2 probe-derived disruption signals yields the largest incremental gain in judged quality, reaching a composite 3.89/5 (mechanism coverage 3.15; biological accuracy 4.27; specificity 4.15) with the strongest interpreter model tier reported.
10. The resource component matters: EVEE provides precomputed predictions and disruption profiles for 4.2 million ClinVar variants, plus on-demand explanations through an interactive explorer—positioning foundation-model embeddings as a shared substrate for both prediction and mechanistic inspection rather than a score-only black box.
💻Code: github.com/goodfire-ai/evee-…
📜Paper: biorxiv.org/content/10.64898…
#ComputationalGenomics #VariantEffectPrediction #GenomicFoundationModels #ClinVar #Interpretability #DeepLearning #Bioinformatics #GenomicMedicine #Splicing #Indels
4
33
1,892
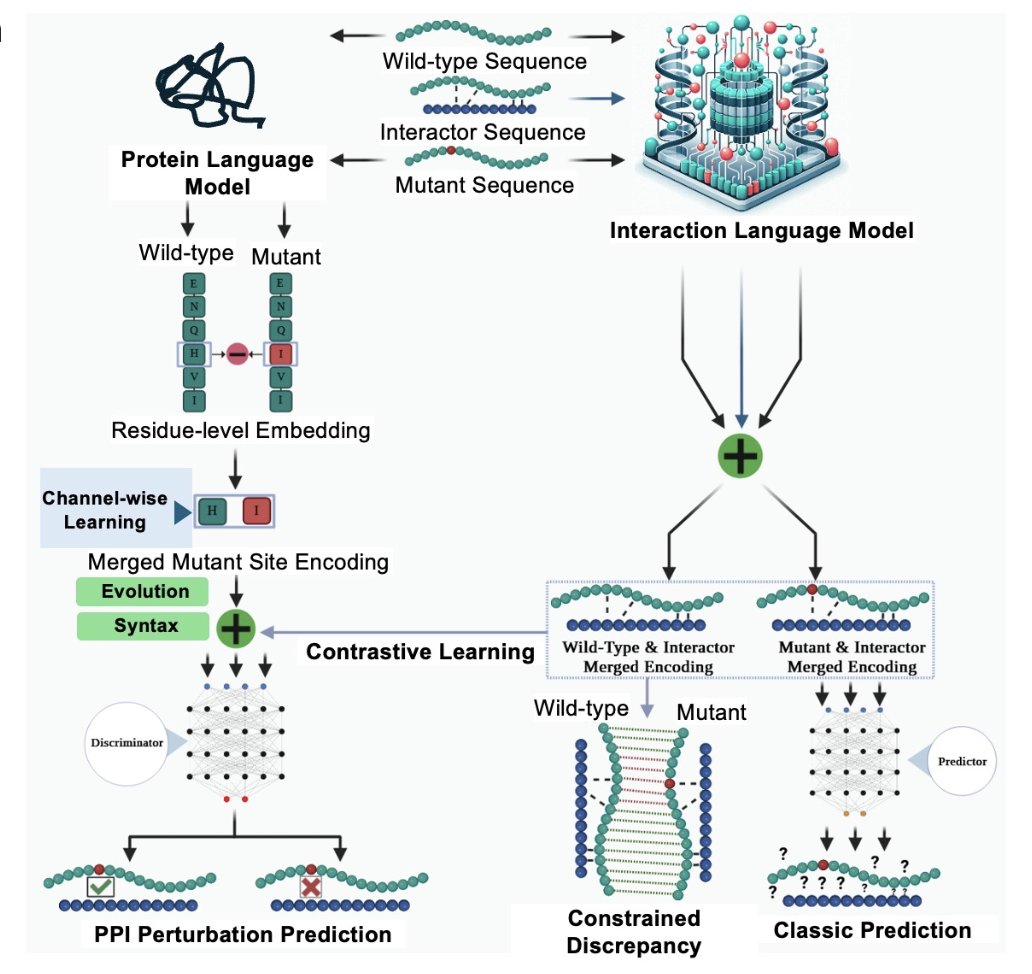
eSIG-Net: Accurate prediction of single-mutation induced perturbations on protein interactions using a language model
1. eSIG-Net frames missense effects as an “interaction cliff” problem: WT and mutant sequences are nearly identical, yet can trigger large, partner-specific PPI rewiring. The model is built to predict interaction perturbations directly (edgetic effects), not just generic PPI presence/absence.
2. Core idea: a sequence-only “interaction language model” that takes (WT protein, mutant protein, interactor) triplets and learns the discrepancy between WT–interactor and mutant–interactor interaction representations, rather than assuming WT and mutant embeddings behave similarly.
3. Architecture highlights two complementary encoders: (i) a PPI protein encoder that embeds WT–interactor and mutant–interactor pairs, and (ii) a mutation-site language-model encoder that extracts residue-level embeddings only at the mutated position (ESM-2), reducing redundancy from whole-sequence pooling.
4. Key innovation is constrained discrepancy learning: the model is explicitly trained so that embedding distances between WT-pair and mutant-pair are small when no perturbation occurs and larger when perturbation occurs, while avoiding trivial collapse via joint training with PPI objectives and a perturbation discriminator.
5. Input feature fusion for PPI encoding uses lightweight, sequence-derived descriptors (AAC 20D Conjoint Triad 343D Auto Covariance 210D = 573D), while mutation-site encoding uses the 1280D ESM-2 residue embedding at the mutation position with channel-wise learning MLP.
6. Benchmark on Sahni et al. disease-mutation PPI dataset (527 mutations; 606 perturbed vs 1,027 non-perturbed PPIs): eSIG-Net reaches accuracy 0.85 vs best competing sequence-based PPI method 0.63; AUC 0.91; average precision 0.86. Competing methods included SDNN, D-SCRIPT, DeepFE, PIPR, and PLM-interact (adapted by comparing WT-pair vs mutant-pair predictions).
7. Robustness on Fragoza et al. population-variant dataset (gnomAD; strongly imbalanced, ~16% positives): eSIG-Net maintains accuracy 0.90 (best other method 0.86), with AUC 0.96 and average precision 0.92—highlighting utility for VUS-like imbalanced settings.
8. Interpretability via latent-space separation: t-SNE visualizations of final-layer representations show eSIG-Net produces clearer separation of perturbed vs non-perturbed cases than baselines (quantified by separation ratio and silhouette score), consistent with explicitly learning WT/MT discrepancies.
9. Comparisons to structure-centric approaches show why “sequence-only but discrepancy-aware” can win for this task: FoldDock (AlphaFold-based docking) predicts WT interactions well but largely outputs the same WT vs mutant decision (94.4% unchanged), failing to detect subtle missense-driven perturbations; structure-based mutation tools (MutaBind2, BeAtMuSiC, GeoPPI, TopNetTree, PIONEER using AlphaFold-Multimer complexes) sit around/below ~60% accuracy on the disease dataset, far below eSIG-Net.
10. Biological case studies illustrate interaction-specific predictions: in TPM3 pleiotropy, eSIG-Net predicts L100M selectively perturbs TPM3–HSF2 while M9R retains it; for COQ8A, disease mutation G272V is predicted to disrupt interactions with RABAC1, REEP6, and TMEM159 whereas population variant H85Q preserves them. Predicted mutation-mediated PPI pairs also show enrichment for clinical associations across TCGA/MMRF, including prognosis and immunotherapy-response correlations.
💻Code: github.com/Yilab-texas/eSIG-…
📜Paper: biorxiv.org/content/10.64898…
#ComputationalBiology #Bioinformatics #ProteinInteractions #PPI #MachineLearning #DeepLearning #ProteinLanguageModels #VariantEffectPrediction #MissenseVariants #Interactome
1
1
13
1,213
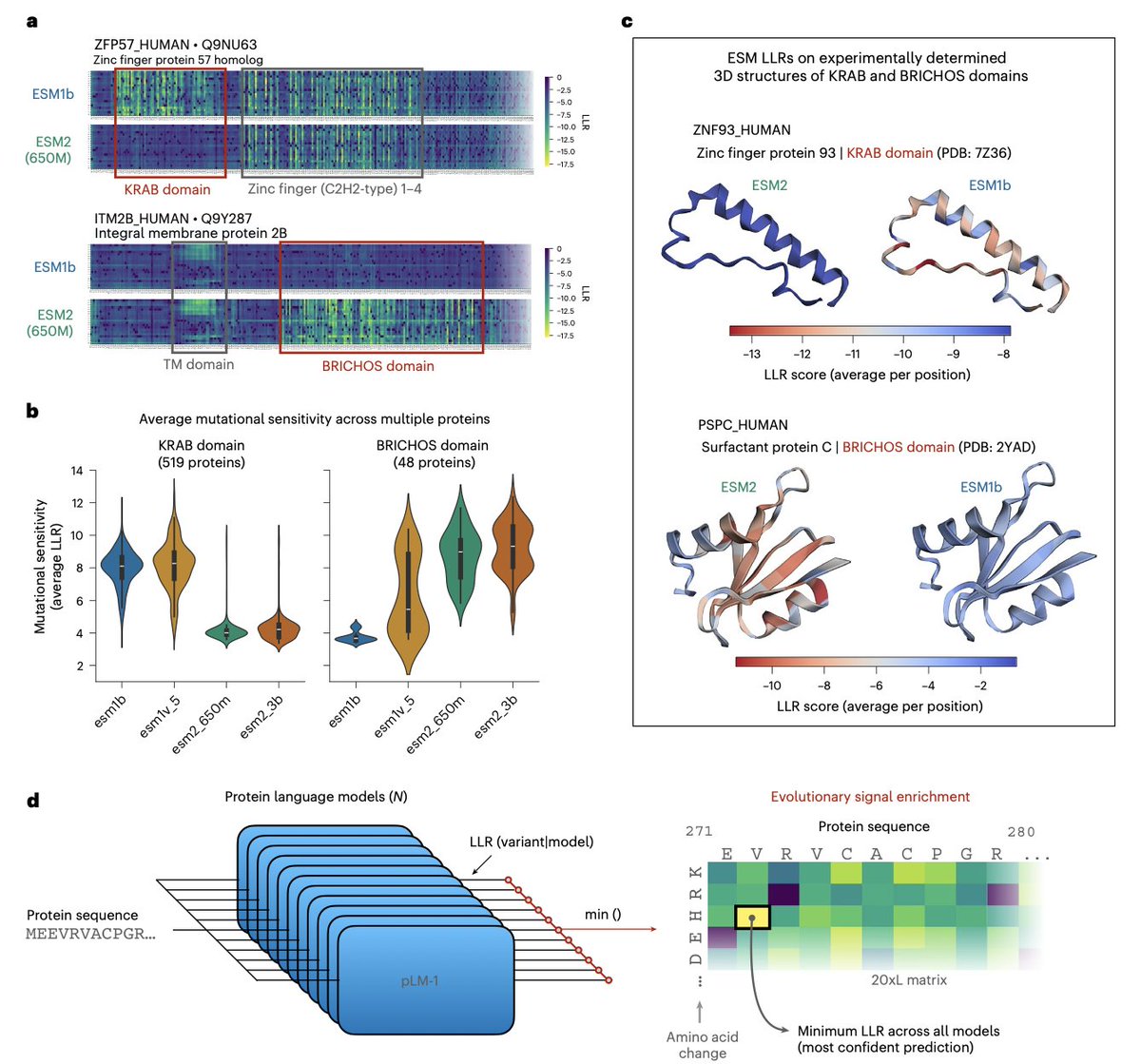
Compressing the collective knowledge of ESM into a single protein language model @naturemethods
1. The paper argues that “sequence-only” protein language models (PLMs) are not intrinsically capped for variant-effect prediction (VEP); instead, their evolutionary signals are fragmented across model families and can be recovered by making models learn from each other.
2. Key observation: closely related ESM models have complementary blind spots. For example, ESM2 models systematically miss KRAB-domain conservation signals, while ESM1b/ESM1v can miss BRICHOS-domain signals; yet at least one model in the family captures each domain’s mutational sensitivity.
3. They introduce a simple but effective ensemble rule: for each missense mutation, take the minimum log-likelihood ratio (LLR) across models (ESMIN), i.e., “maximum confidence” scoring. This can amplify subtle evolutionary constraints that averaging would dilute.
4. A theoretical analysis explains when min-LLR beats averaging: if pathogenic-variant LLRs are more dispersed across models than benign-variant LLRs (variance asymmetry). The ESM family empirically shows this property, making maximum-confidence aggregation advantageous.
5. ESMIN is evaluated using 11 sequence-only ESM models (ESM1b, five ESM1v, five ESM2; excluding ESM2-15B). It outperforms averaging-based ensembles and improves ProteinGym DMS correlations, with gains occurring in ~50% of assays (versus ~20% for typical ensembles).
6. Main methodological contribution: “maximum-confidence co-distillation.” For each protein, all models score all mutations; the element-wise minimum LLR matrix becomes a teacher signal, and each model is trained (variant-level MSE) to match these confident targets—without MSAs, structures, or population genetics features.
7. Co-distillation substantially improves every participating model, including small ones: ESM2-8M improves on ClinVar AUC from ~0.65 to ~0.88. Several co-distilled single models (e.g., ESM2-3B, ESM1b, ESM2-650M) can even surpass the ESMIN teacher signal (“student surpasses teacher”).
8. Robustness/ablation: improvements persist when training data are heavily reduced and de-homologized. With only ~1% of human proteins (~200 sequences; <30% identity to benchmark proteins), ESM2-35M reaches ~97% (ClinVar) and ~94% (DMS) of its peak co-distilled performance.
9. Iterative procedure: after round 1 (min-LLR co-distillation), additional rounds switch to average-aggregation co-distillation. As models improve, class-conditional variances become more symmetric, making averaging slightly better; after 3 rounds, a single 3B model matches the ensemble—named VESM-3B.
10. Practical compression: VESM-3B is distilled into smaller models (650M, 150M, 35M) that retain most performance (reported as >98% on Balanced ClinVar and >93% on ProteinGym DMS relative to VESM-3B), enabling high-throughput VEP under limited compute.
11. Clinical benchmark (ProteinGym ClinVar, 2,227 genes): sequence-only VESM models outperform other sequence-only PLMs (including ESM-C) and compete with or surpass methods using MSA/structure/population priors. VESM-3B shows balanced ROC behavior across specificity and sensitivity regimes.
12. AlphaMissense comparison: VESM-3B performance is stable across allele-frequency strata, while AlphaMissense shows strong dependence on MAF (consistent with circularity risks when population frequency informs clinical labels). After excluding variants overlapping AlphaMissense training (gnomAD v2 MAF > 1e-5), all VESM sizes outperform AlphaMissense on AUC and multiple calibrated metrics.
13. Modular use of structure: rather than retraining a joint model, they fine-tune the sequence component of ESM3 using VESM-style sequence-based loss to create VESM3, and combine VESM3 with VESM-3B into a structure-aware ensemble (VESM ). This improves performance on structure-dependent DMS assays (binding/stability/expression) while maintaining strong fitness/activity performance.
14. Cross-domain generalization: despite co-distillation being trained on human proteins, gains transfer strongly to nonhuman DMS assays, with disproportionately large improvements reported for viral proteins—even though ESM3’s released training data excluded viral sequences.
15. Beyond binary pathogenicity: using UK Biobank/Genebass summary statistics for 332 gene–phenotype pairs (blood biochemistry biomarkers), variant-level VESM scores correlate with single-variant effect sizes (β). VESM and VESM-3B yield the strongest gene–trait association signals across tested models.
16. Notably, VESM-3B recovers the correct pLoF direction of effect in 98.8% of significant gene–phenotype pairs and identifies many associations not detected by missense burden tests, suggesting utility for quantitative trait interpretation from summary statistics.
📜Paper: doi.org/10.1038/s41592-026-0…
#ProteinLanguageModels #VariantEffectPrediction #ComputationalBiology #HumanGenetics #ESM #ClinVar #ProteinGym #DeepMutationalScanning #UKBiobank #MachineLearning
1
29
125
22,027
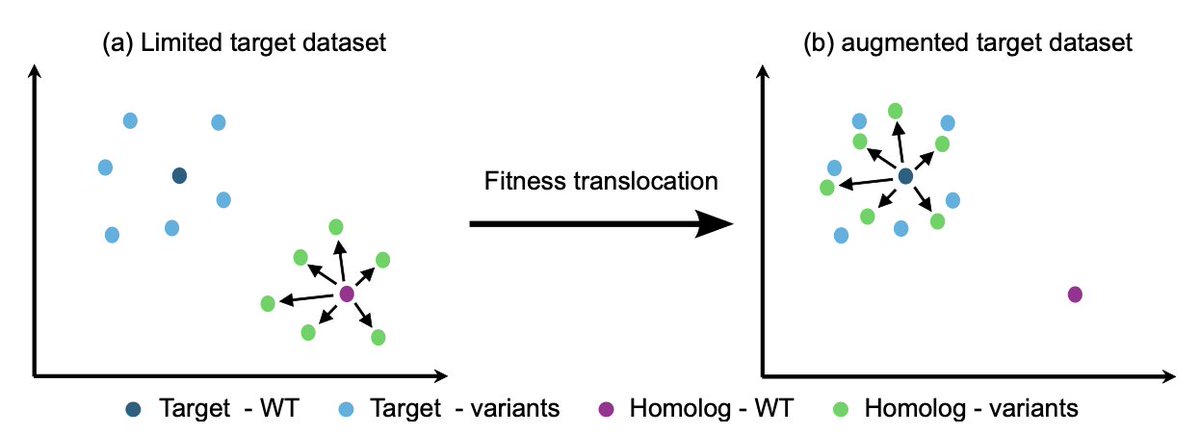
Fitness Translocation: Improving Variant Effect Prediction with Biologically-Grounded Data Augmentation
1. The study tackles the long‑standing bottleneck of sparse protein fitness data, which hampers accurate mapping from sequence to function in engineering and evolution.
2. It introduces *fitness translocation*, a data‑augmentation strategy that transfers the mutational effect observed in a homologous protein to a target protein, thereby enriching the target’s training set without new experiments.
3. Using pretrained protein language models, the method computes an embedding offset for each homolog variant (variant embedding minus wild‑type embedding) that captures the mutation’s directional change in latent space.
4. The offset is applied to the target wild‑type embedding, creating a synthetic variant that inherits the homolog’s measured fitness (normalized by wild‑type fitness), thus preserving biological relevance in the augmented data.
5. Because the technique operates solely in embedding space, it requires no sequence alignment and can be applied to homologs with as little as 35 % sequence identity, making it well suited for low‑data regimes.
6. The authors benchmarked fitness translocation on three diverse protein families—IGPS enzymes, green fluorescent proteins, and SARS‑CoV‑2 spike proteins—using multiple predictors (SVR, RF, Lasso) and language models (ESM‑2, ESM‑1v).
7. Across all configurations, augmentation consistently improved Spearman correlation, with the largest gains observed for SARS‑CoV‑2 spike cell‑entry predictions, followed by IGPS enzymatic activity and GFP fluorescence.
8. A homolog‑selection algorithm, grounded in one‑sided paired t‑tests, identifies which homologs yield statistically significant performance boosts, preventing the inclusion of noisy or irrelevant data.
9. Ablation studies show that removing either the statistical test or the sequential selection stage degrades results, underscoring the algorithm’s role in achieving robust improvements.
10. Principal‑component analysis demonstrates that translocation aggregates homolog variant embeddings around the target, indicating that mutational impacts are effectively transferred across sequence space.
11. The method’s success aligns with evidence that fitness landscapes are conserved across phylogenetically distant proteins, validating the biological assumption that evolutionary pressures preserve functional constraints.
12. By expanding usable training data, fitness translocation can accelerate directed evolution and generative protein design, potentially reducing the number of costly experimental cycles needed to reach high‑performance variants.
💻Code: github.com/adrienmialland/Pr…
📜Paper: biorxiv.org/content/10.1101/…
#ProteinEngineering #MachineLearning #ProteinLanguageModels #VariantEffectPrediction #DataAugmentation #DirectedEvolution #ComputationalBiology #SARSCoV2 #IGPS #GFP #Bioinformatics
3
13
1,574
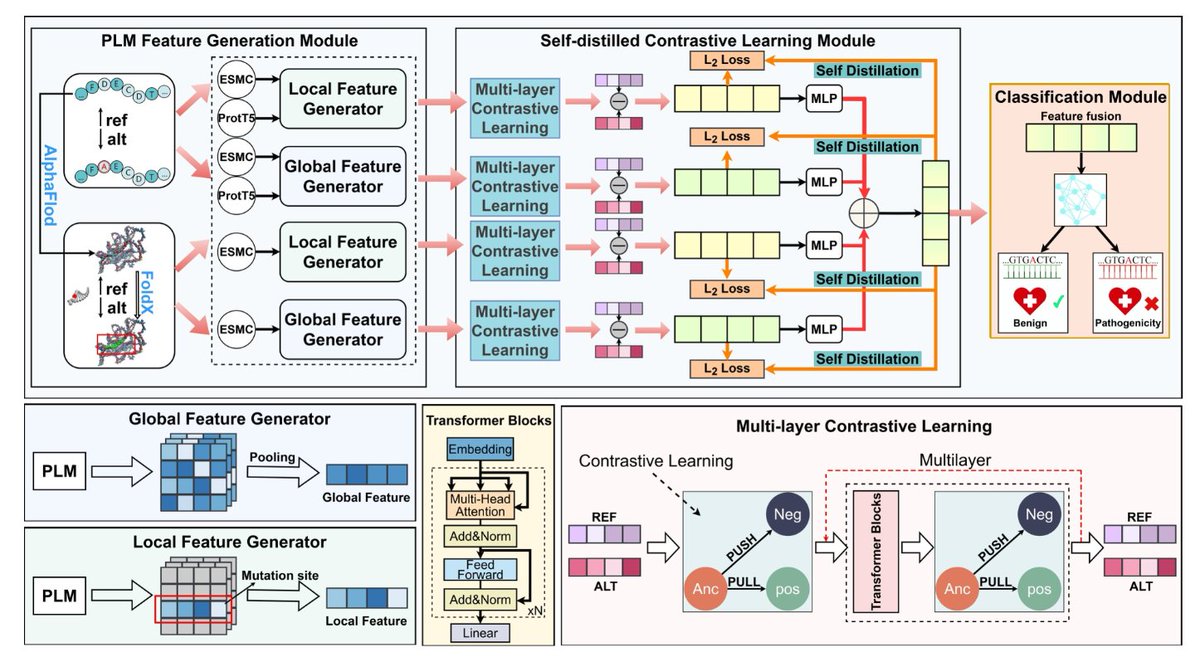
VarDCL: A Multimodal PLM-Enhanced Framework for Missense Variant Effect Prediction via Self-distilled Contrastive Learning
1 The VarDCL framework fuses protein sequence and structure embeddings from state‑of‑the‑art language models (ProtT5 and ESMC) to capture both global and local mutation context, providing a richer multimodal input than prior single‑modal predictors.
2 A novel Self‑Distilled Contrastive Learning (SDCL) module is introduced, comprising multi‑level contrastive learning that aligns wild‑type and mutant representations at multiple feature depths, and a self‑distillation mechanism that transfers high‑level fused knowledge to low‑level differential features, enhancing cross‑modal interaction.
3 By jointly optimizing binary cross‑entropy with the SDCL objective, VarDCL learns highly discriminative representations that are sensitive to subtle sequence and structural changes induced by missense variants.
4 Extensive ablation studies confirm that both the multi‑level contrastive component and the self‑distillation module are essential; removing either reduces AUC by 1–2% and MCC by over 4%, underscoring their synergistic effect.
5 On an independent test set of 18,731 clinical variants, VarDCL attains an AUC of 0.917, an AUPR of 0.876, an MCC of 0.690, and an F1 score of 0.789, surpassing 21 existing state‑of‑the‑art variant effect predictors.
6 The model’s superior performance is attributed to its ability to jointly model dynamic pre‑ and post‑mutation changes and to facilitate deep information exchange between sequence and structural modalities, setting a new benchmark for missense variant pathogenicity prediction.
💻Paper: biorxiv.org/content/10.64898…
#ProteinEngineering #Genomics #MachineLearning #VariantEffectPrediction #Bioinformatics
2
13
1,109
Benchmarking DNA Foundation Models: Biological Blind Spots in Evo2 Variant-Effect Prediction
1. This preprint presents a rigorous controlled evaluation of Evo2, a large DNA foundation model, revealing critical biological blind spots that challenge claims of zero-shot pathogenicity prediction readiness.
2. The authors developed a comprehensive benchmarking framework testing short-range (codon usage, mitochondrial codon idiosyncrasies), medium-range (tRNA context sensitivity), and long-range signals (gene completion, NUMT disambiguation, evolutionary conservation).
3. A striking finding: Evo2 fails to capture human codon usage bias, with wobble-base predictions near-random (preferred codon selected only 24.4% of the time, mean JSD = 0.254) despite training on 9 trillion bases from 16,000 eukaryotic genomes.
4. The tRNA permutation experiment demonstrates spurious context sensitivity—when mitochondrial tRNAs were cyclically relocated while keeping sequences intact, pathogenicity prediction sensitivity collapsed from 65.8% to 5.1%, proving predictions are driven by flanking sequence rather than tRNA structure itself.
5. Evo2 misclassifies most valid mitochondrial start/stop codon alternatives as pathogenic (100% of start codon variants, 72.7% of stop codon variants), showing it applies nuclear genetic code conventions to mtDNA.
6. While Evo2 achieves competitive aggregate metrics (AUROC 0.896, MCC 0.631), performance varies dramatically by region: 100% accuracy on mild pathogenic variants but worse on severe ones—a clinically concerning inversion.
7. The NUMT analysis reveals Evo2 defaults to mitochondrial reference alleles at divergence sites, treating nuclear mitochondrial pseudogenes as authentic mtDNA regardless of nuclear context provided.
8. Gene completion accuracy does not track biological constraint: Complex III, the most mutation-intolerant OXPHOS complex, shows the lowest completion accuracy (85.0%), opposite to biological expectations.
9. These findings suggest that unsupervised scaling on raw sequence alone is insufficient for clinical-grade variant interpretation, and that hierarchical biological supervision is needed for trustworthy genomic foundation models.
📜Paper: biorxiv.org/content/10.64898…
#Genomics #Bioinformatics #MachineLearning #VariantEffectPrediction #MitochondrialDNA #FoundationModels #Evo2 #ComputationalBiology #ClinicalGenomics #DNALanguageModels

1
10
43
4,913
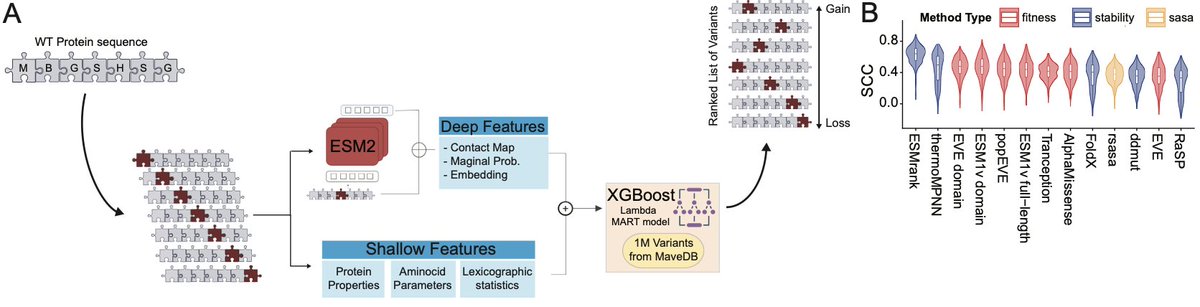
ESMRank Reveals a Transferable Axis of Protein Mutational Constraint from Overlapping Variant Effect Assays
1. A new study introduces ESMRank, a learning-to-rank framework that outperforms existing stability predictors by leveraging the ordinal structure hidden in overlapping multiplexed assays of variant effect (MAVEs).
2. The key insight: while absolute effect sizes vary wildly across different experiments, the relative ranking of variants within proteins is remarkably consistent. The authors exploit this redundancy to build a unified "variant soundness" metric.
3. The method integrates ~1.1 million variants from MAVEdb using Reciprocal Rank Fusion, revealing that buried residues and packing perturbations dominate the constraint landscape across heterogeneous assays.
4. ESMRank combines ESM-2 protein language model embeddings with physicochemical descriptors in a LambdaMART ranking model, achieving state-of-the-art performance on Human Domainome and ProteinGym stability benchmarks without structure input.
5. Remarkably, the model stratifies disease genes by mechanism—haploinsufficient genes show highest constraint, gain-of-function genes lowest—despite never seeing clinical labels during training.
6. In a detailed CFTR case study, ESMRank scores correlate with folding efficiency (ρ=0.56), channel activity (ρ=0.65), and critically, pharmacological rescue potential, distinguishing gating- from processing-dominant variants.
7. The work establishes experimental overlap as a scalable statistical resource and demonstrates that aligning machine learning objectives with intrinsic biological ordering yields more transferable predictions than regression approaches.
💻Code: github.com/arneseric/esmrank
📜Paper: biorxiv.org/content/10.64898…
#ProteinEngineering #VariantEffectPrediction #DeepMutationalScanning #ProteinLanguageModels #ComputationalBiology #Bioinformatics #MachineLearning #ProteinStability #PrecisionMedicine
4
27
1,891
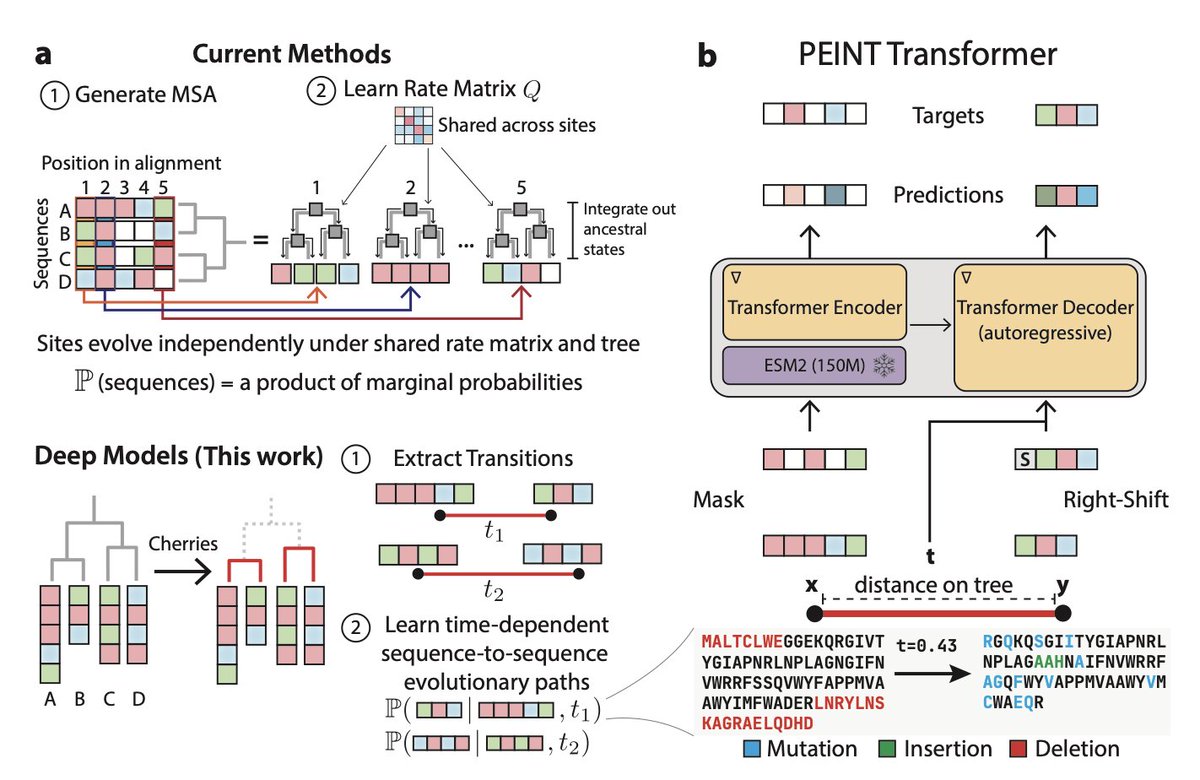
Deep Models of Protein Evolution in Time Generate Realistic Evolutionary Trajectories and Functional Proteins
1 PEINT (Protein Evolution IN Time) introduces a deep learning framework that models how entire protein sequences evolve over time while capturing complex interactions between sites, moving beyond the biologically unrealistic independent-sites assumption that has dominated phylogenetics for decades.
2 The model uses a coupled encoder-decoder transformer architecture built on top of ESM2, enabling it to learn directly from raw unaligned sequences without requiring pre-computed multiple sequence alignments, thereby eliminating alignment-induced biases.
3 PEINT learns insertion-deletion (indel) dynamics natively from sequence data, addressing a major limitation of classical models that typically ignore these critical evolutionary processes despite their outsized importance in protein evolution.
4 When simulating evolution along phylogenetic trees, PEINT generates highly novel sequences with only ~50% identity to any known protein while preserving structural integrity and function, as experimentally validated through functional characterization of designed carbonic anhydrase variants.
5 The model demonstrates superior performance in likelihood-based evaluation of evolutionary transitions, accurate inference of branch lengths from unaligned sequences (Pearson's r=0.95 with classical methods), and generation of sequences with high structural confidence scores even at high mutational loads.
6 PEINT improves variant effect prediction over base protein language models, particularly for organismal fitness assays and variants with multiple mutations, by leveraging family-specific evolutionary information through a principled training objective.
7 Mechanistic interpretability reveals that PEINT's decoder learns to align target sequences to source sequence representations during generation, recovering ground truth alignments with accuracy comparable to dynamic programming algorithms despite no explicit training for alignment.
8 This work establishes a foundation for realistic simulation of protein evolution that balances exploration of new sequence space with respect for structural and functional constraints, offering powerful tools for phylogenetic inference and protein engineering.
📜Paper: biorxiv.org/content/10.64898…
#ProteinEvolution #DeepLearning #Phylogenetics #ProteinDesign #ComputationalBiology #MachineLearning #Bioinformatics #VariantEffectPrediction #ProteinLanguageModels
4
28
1,841
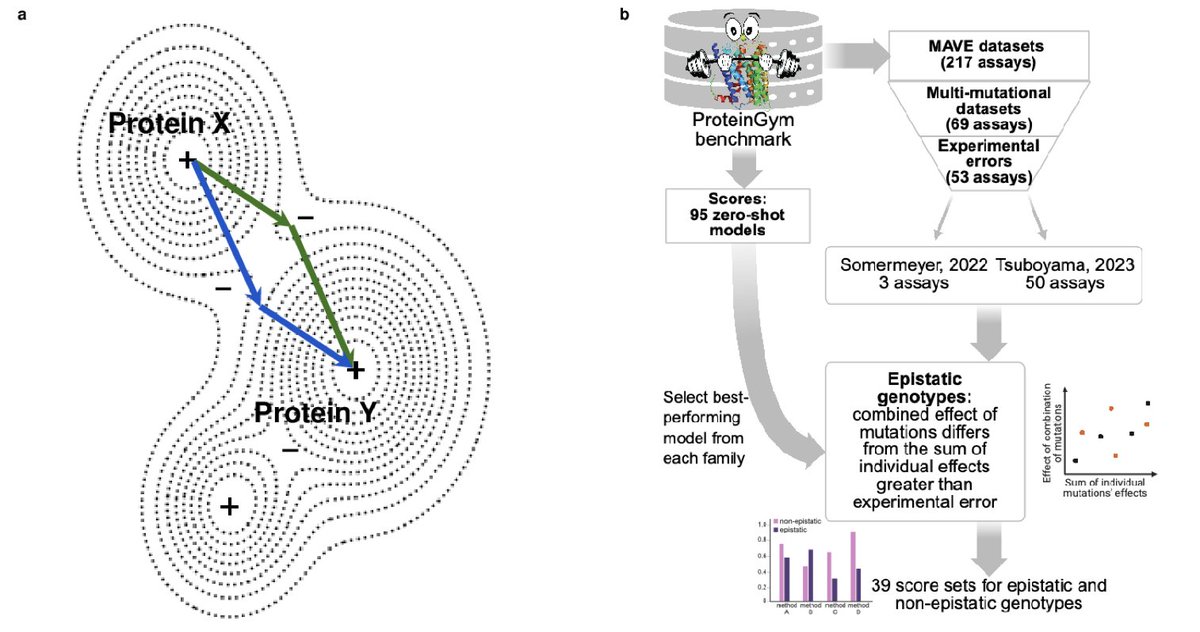
Beyond additivity: zero-shot methods cannot predict impact of epistasis on protein properties and function
1 The study reveals a critical blind spot in modern protein AI: while 95 state-of-the-art zero-shot models can predict single mutations well, they systematically fail when mutations interact epistatically—where the combined effect of mutations deviates from simple additivity.
2 Using 53 MAVE datasets from ProteinGym, the researchers identified epistatic genotypes by comparing observed effects against expected additive effects, accounting for experimental error. For GFP fluorescence and protein thermostability, epistasis is widespread and biologically genuine, not a measurement artifact.
3 The performance gap is stark. Top models like ESCOTT, PoET, and MSA-Transformer achieve Spearman correlations above 0.6 for all genotypes, but collapse to near-zero or negative correlations for epistatic genotypes. Simple linear regression baselines often match or exceed complex deep learning models on epistatic combinations.
4 This exposes a fundamental limitation: protein language models learn evolutionary plausibility from natural sequences, but natural selection only explores functional sequence space. Epistatic combinations—often traversing fitness valleys—lie outside this training distribution, leaving models blind to higher-order mutational interactions.
5 The work highlights that clever feature engineering (evolutionary conservation, structural information) outperforms architectural complexity for epistasis prediction. Yet even structure-aware models like ProSST and ESM-IF1, while top performers on stability, show no consistent advantage across datasets.
6 The implications are profound for protein design and directed evolution. Current zero-shot methods cannot reliably navigate rugged fitness landscapes or predict functional variants along evolutionary paths requiring epistatic mutations. The field urgently needs models trained on multi-mutational data and architectures explicitly modeling non-linear interactions.
💻Code: github.com/kalininalab/epist…
📜Paper: biorxiv.org/content/10.64898…
#ProteinEngineering #Epistasis #MachineLearning #ProteinGym #VariantEffectPrediction #ComputationalBiology #Bioinformatics #ProteinEvolution #AIforScience #StructuralBiology
2
38
11,773
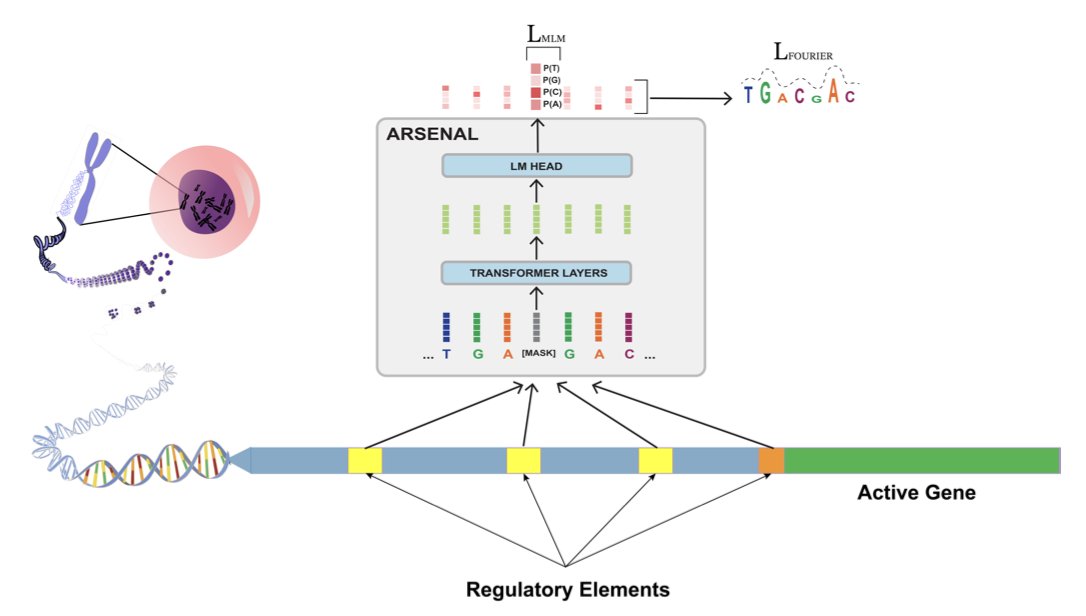
Short-Context Regulatory DNA Language Models with Motif-Discovery Regularization
1 ARSENAL is a new DNA language model specifically designed to learn regulatory sequence syntax, addressing a key limitation of existing whole-genome models that struggle with sparse, motif-scale regulatory features.
2 The model introduces two core innovations: training on ENCODE candidate cis-regulatory elements (cCREs) rather than entire genomes, and a novel Fourier transform-based regularizer that explicitly encourages discovery of short (6-20bp) transcription factor binding motifs.
3 Unlike massive long-context models, ARSENAL uses a compact 350bp context window, demonstrating that targeted training with biological priors can outperform scale alone for regulatory tasks.
4 In zero-shot evaluations, ARSENAL substantially outperforms existing DNA language models on motif identification and variant effect prediction, achieving Pearson correlations of ~0.4 on dsQTL benchmarks where other models score near zero.
5 The Fourier regularizer proves critical—an ablation without it shows dramatically degraded performance, confirming that raw self-supervision on regulatory regions alone is insufficient for learning meaningful motif syntax.
6 ARSENAL's embeddings improve supervised chromatin accessibility prediction across multiple cell types when incorporated into ChromBPNet-style models, with consistent gains in GM12878, K562, HepG2, IMR90, and H1-hESC cell lines.
7 The model enables practical regulatory sequence design through beam search optimization, generating sequences with targeted accessibility levels and cell-type specificity while maintaining interpretability through discovered motif patterns.
8 For cell-type-specific design, optimized sequences correctly enrich for lineage-appropriate motifs—FOX/HNF/C/EBP for HepG2 and OCT4/SOX2 for H1-hESC—demonstrating that the model captures biologically meaningful regulatory grammar.
💻Code: github.com/kundajelab/regula…
📜Paper: biorxiv.org/content/10.64898…
#DNALM #RegulatoryGenomics #DeepLearning #MotifDiscovery #VariantEffectPrediction #ChromatinAccessibility #SyntheticBiology #Genomics #Bioinformatics
11
36
3,829
Predicting Evolutionary Rate as a Pretraining Task Improves Genome Language Model Representations
1) The paper introduces a novel approach to training genome language models (gLMs) by predicting evolutionary rates as a pretraining task, moving beyond traditional sequence reconstruction methods that often fail to capture meaningful biological signals.
2) Two novel pretraining tasks are proposed: Current Evolution Prediction (CEP), which predicts evolutionary rates autoregressively like next token prediction, and Masked Evolution Modeling (MEM), which predicts rates using bidirectional context like masked language modeling.
3) These evolutionary prediction tasks can be seamlessly composed with existing sequence reconstruction tasks (NTP and MLM), enabling controlled comparisons between training on sequence only, evolution only, or both combined.
4) The authors developed "Gamba" models (ArGamba and Bi-Gamba) ranging from 4M to 66M parameters, demonstrating that relatively small models pretrained on evolutionary rates can compete with much larger existing gLMs like Evo2 (7B parameters) on several benchmarks.
5) A comprehensive suite of biologically grounded zero-shot benchmarks was created to evaluate model representations, including distinguishing functional genomic regions under increasing difficulty settings and classifying ATG codons in different functional contexts.
6) Key finding: Models pretrained on both sequence and evolutionary rate consistently outperform sequence-only models across most tasks, with evolutionary rate-only models also showing strong performance, establishing evolution as a critical training signal for genome-scale models.
7) The work addresses major gaps in gLM evaluation by measuring zero-shot performance without supervised classifiers, including randomly-initialized baselines, and using multiple negative sets of systematically increasing difficulty to reveal what signals models actually learn.
8) On variant effect prediction benchmarks, evolutionary rate prediction provides substantially stronger signal than sequence likelihood alone, with Gamba models matching or outperforming Evo2 on several tasks despite being orders of magnitude smaller.
💻Code: github.com/macaes-lab/gamba
📜Paper: biorxiv.org/content/10.64898…
#Genomics #Bioinformatics #MachineLearning #ComputationalBiology #GenomeLanguageModels #EvolutionaryGenomics #DeepLearning #Pretraining #VariantEffectPrediction
2
38
2,386
Functional In-Context Learning in Genomic Language Models with Nucleotide-Level Supervision and Genome Compression
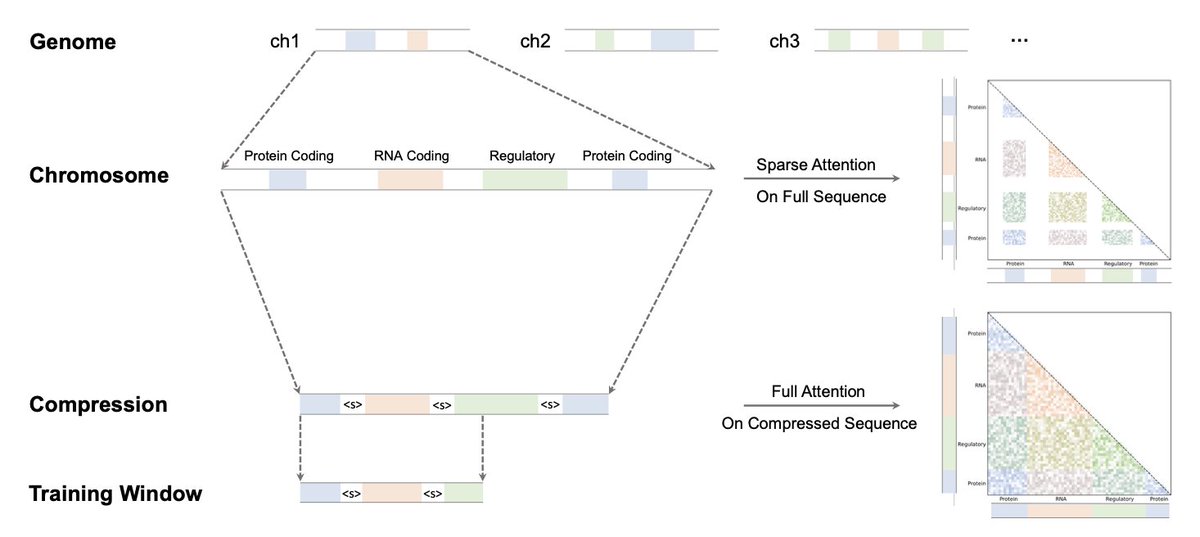
1. GENERator-v2 keeps the vanilla Transformer and 6-mer tokenization, yet pushes SOTA on 98 kbp contexts by re-designing the loss and the data instead of the architecture.
2. Factorized Nucleotide Supervision (FNS) turns one 4^6-way k-mer prediction into six independent nucleotide likelihoods via simple marginalisation, restoring single-base resolution without extra compute.
3. Genome Compression Pre-training (GCP) concatenates gene-centric and regulatory intervals, discarding megabases of low-signal DNA; the “compressed genome” lets a dense attention window cover dozens of genes in one pass.
4. The combo yields explicit next-gene prediction, giving the model a built-in sense of functional neighbourhood and long-range co-regulation.
5. On 30-nt sequence recovery GENERator-v2-1B outperforms Evo2-1B and matches Evo2-7B while running 20× faster; the 3B variant sustains gains up to 36 kbp input.
6. Variant effect prediction on ClinVar reaches AUPRC 0.942—identical to Evo2-7B—using half the parameters and the same wall-clock time.
7. Separator-token embeddings learned under GCP collapse codon phase and strand orientation, clustering sequences by species, a direct representation-level signature of functional in-context learning.
8. Training-free evaluations and 18 revised NT fine-tuning tasks both improve over GENERator-v1, indicating that better supervision and data trump architectural tinkering.
9. Models and inference code are released for eukaryotic and prokaryotic genomes, offering an efficient drop-in replacement for long-context genomic generation and SNP scoring.
💻Code: huggingface.co/GenerTeam
📜Paper: biorxiv.org/content/10.64898…
#GenomicFoundationModel #LongContextDNA #NucleotideSupervision #GeneCompression #VariantEffectPrediction #bioinformatics
7
14
1,586
When Less Is Enough: Low-Rank Structure in DNA Sequence-to-Function Models
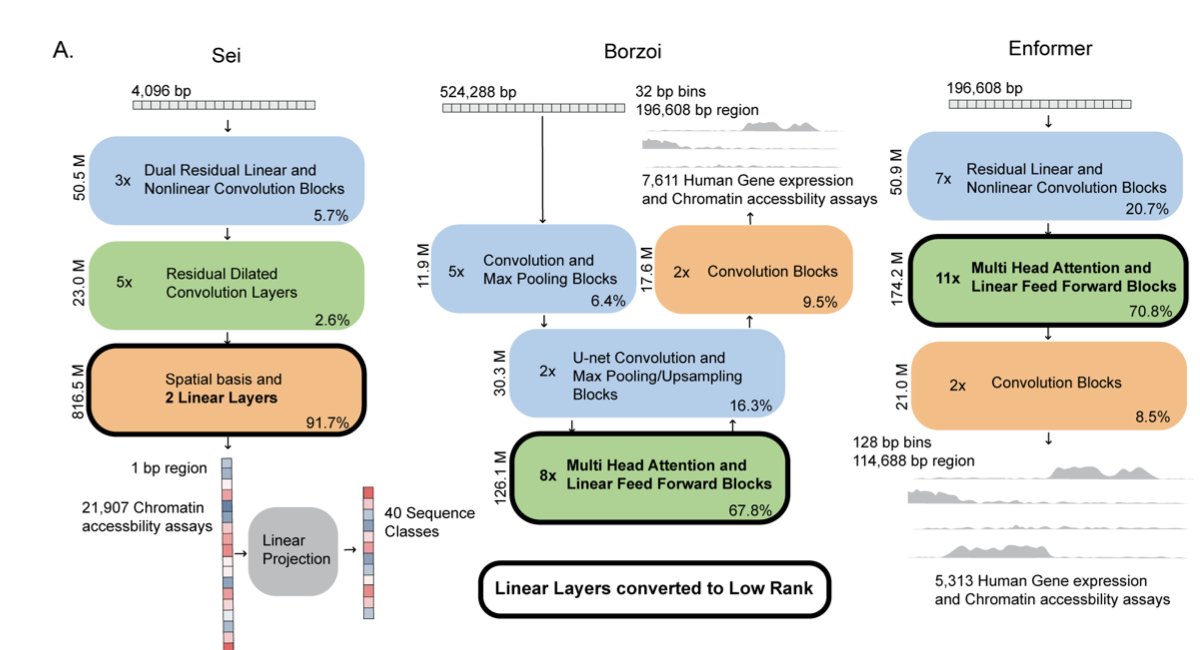
1. A new preprint by Gilfeather, Chikina & Kostka shows that 90 % of parameters in large regulatory-genomics deep nets can be tossed without hurting accuracy.
2. Trick: post-hoc singular-value decomposition of every linear layer in Sei, Borzoi & Enformer yields “LLRA” models whose weight matrices are rank-k with k as low as 1.
3. Shockingly, rank-1 Sei-LLRA beats the full 890 M-parameter model on most promoter-variant benchmarks, hinting that chromatin-state signals live in a tiny linear subspace.
4. Combine low-rank weights with 8-bit static quantization → 5× CPU speed-up and >100× faster per-sequence inference than Borzoi/Enformer while keeping ρ>0.96 with full outputs.
5. On 7 promoter-eQTL & 6 cell-type-specific QTL datasets, quantized Sei-LLRA (rank 64–256) matches or outperforms the parent model and runs on a laptop in seconds.
6. Authors release the “seillra” Python package that lets anyone swap ranks (1–2048) and toggle CPU/GPU inference—no GPU farm required for high-quality variant effect scoring.
7. Work reframes model-compression from an engineering hack to a scientific probe: if rank-1 captures the biology, maybe we should rethink how much complexity regulatory sequence really needs.
💻Code: github.com/kostkalab/seillra
📜Paper: biorxiv.org/content/10.64898…
#Bioinformatics #RegulatoryGenomics #ModelCompression #VariantEffectPrediction #LowRank #CPUfriendly
6
16
1,473

3 Dec 2025
StructGuy: Data leakage free prediction of functional effects of genetic variants
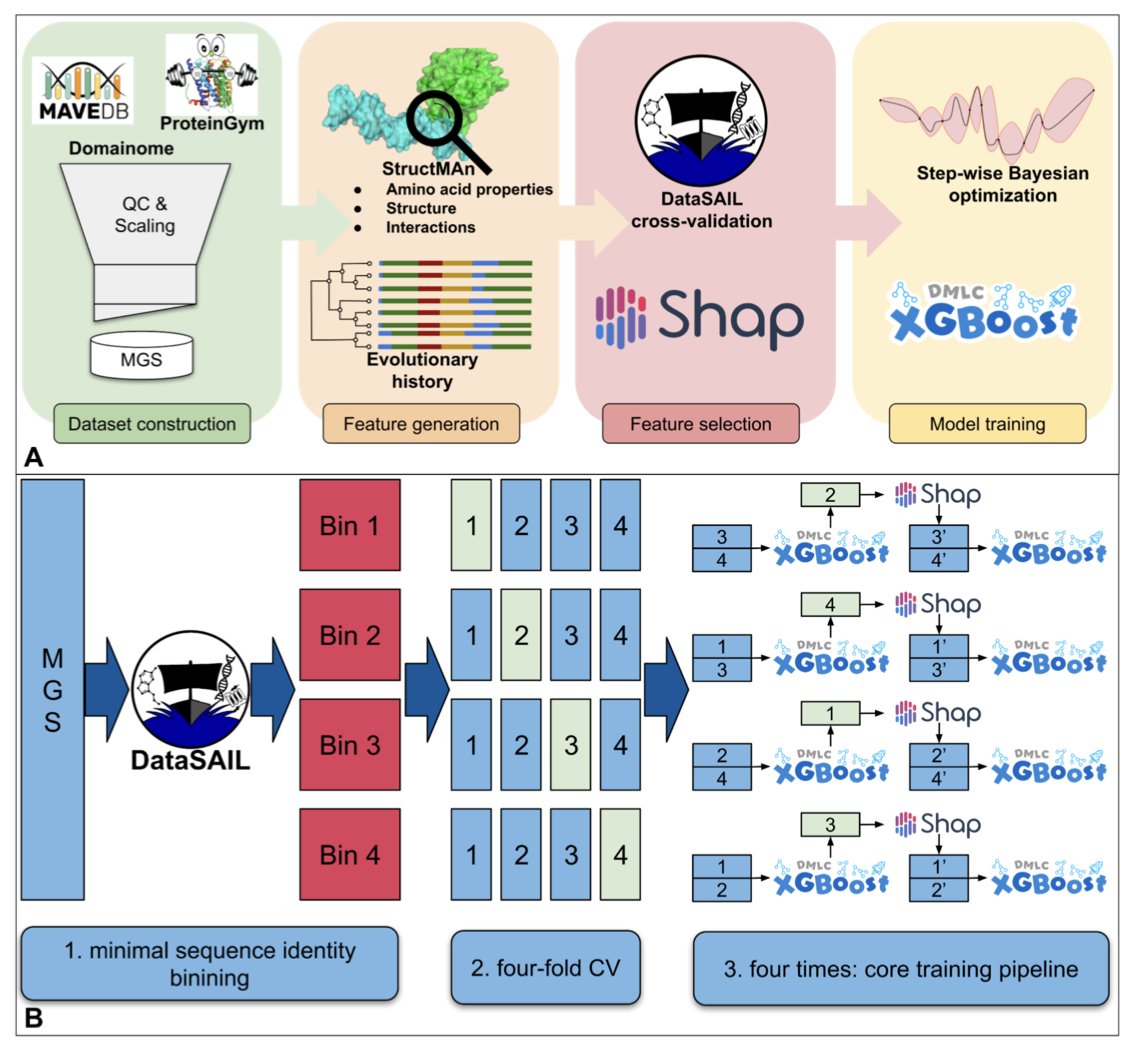
1. A new supervised machine learning model, StructGuy, has been introduced to predict the functional effects of genetic variants on proteins. Unlike other models, StructGuy is designed to generalize predictions to proteins not seen during training, addressing a significant challenge in variant effect prediction.
2. The study leverages a dedicated training dataset derived from multiplexed assays of variant effects (MAVE) experiments. This dataset is carefully curated to prevent data leakage, ensuring that the model can accurately predict effects on unseen proteins.
3. StructGuy employs gradient boosting trees and integrates comprehensive protein structure-based features, including interactions and evolutionary information. This approach allows the model to provide fully interpretable predictions, offering insights into how mutations affect protein 3D structure.
4. The model's performance was evaluated using a modified ProteinGym benchmark, demonstrating competitive accuracy compared to state-of-the-art zero-shot methods. StructGuy's ability to generalize across different proteins makes it a valuable tool for variant effect prediction.
5. In a case study on the peroxisome proliferator-activated receptor gamma (PPARG), StructGuy successfully predicted the functional impact of mutations, illustrating its potential for mechanistic molecular hypotheses. This highlights the model's practical application in understanding variant effects at the protein level.
📜Paper: biorxiv.org/content/10.64898…
#StructGuy #VariantEffectPrediction #MachineLearning #ProteinFunction #Bioinformatics
3
11
1,321
20 Oct 2025
Fine-tuning protein language models on human spatial constraint yields state-of-the-art variant effect prediction
1. A novel study introduces Human Spatial Constraint (HSC), a novel framework that integrates population-scale human genetic variation with 3D protein structures to quantify intraspecies constraint on missense variants. This approach outperforms traditional conservation metrics and unsupervised protein language models in predicting pathogenic variants.
2. The HSC framework maps missense and synonymous variants from hundreds of thousands of individuals onto predicted 3D protein structures. By comparing observed variant frequencies to expected frequencies under neutral evolution, it identifies regions of proteins that are under strong evolutionary constraint.
3. HSC scores highlight functionally important regions of proteins, such as DNA-binding and ligand-binding domains. For example, in the peroxisome proliferator-activated receptor γ (PPARG), HSC scores reveal strong constraints on the interfaces with RXRα and DNA, consistent with known functional regions.
4. The study demonstrates that HSC outperforms other intraspecies and interspecies constraint metrics in predicting pathogenicity. It achieves ROC and PR AUCs of 0.91 and 0.90, respectively, in distinguishing pathogenic from benign missense variants in ClinVar.
5. The impact of spatial context on constraint quantification is explored. The study finds that a 5 Å context window offers the best balance for leveraging structural context without leading to false positives in pathogenicity prediction.
6. Combining HSC with deep learning-based models like AlphaMissense and ESM1b improves pathogenicity prediction. Logistic regression models combining HSCZ_5 with these models achieve ROC AUCs of 0.95, highlighting the complementary nature of HSC.
7. Fine-tuning protein language models (PLMs) using HSC scores improves protein fitness predictions. The fine-tuned ESM2 models show consistent performance gains across various model sizes and DMS datasets, particularly for stability, enzymatic activity, and organismal fitness.
8. The study concludes that integrating intraspecies constraint with cross-species PLMs provides a more comprehensive view of protein function and variant effects. Future work could incorporate adaptive context windows and conformational ensembles to further enhance constraint estimation.
📜Paper: biorxiv.org/content/10.1101/…
#ProteinLanguageModels #HumanSpatialConstraint #VariantEffectPrediction #ComputationalBiology #Genetics

1
7
43
2,850