
Trained a GraphNN on this slutcloud and it told me to take a shower.
Apr 12

Glosso has a slutcloud function, where you can add people you've had sex with and it makes a visualization of all the sex connections between everyone. Here's my slutcloud:

1
2
129
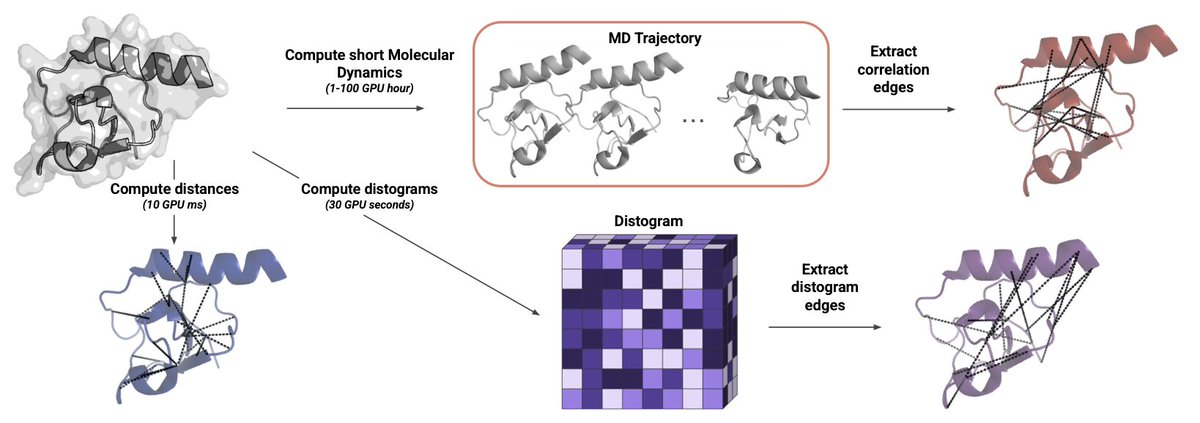
Learning Dynamic Protein Representations at Scale with Distograms
1. The authors bypass expensive MD by mining AlphaFold2/Boltz2 distograms—probability maps of residue–residue distances—to inject true conformational heterogeneity into graph neural networks without generating a single extra structure.
2. They turn each predicted distance distribution into a new edge set (Edisto) plus 64-bin edge features; a relational GNN then learns separate message-passing rules for static 3-D contacts, MD-derived motion correlations, and these cheap distogram links.
3. Across 24 architecture/task combinations distogram edges rank first 16× and second 8×; on ligand-binding-site prediction they raise F1 by up to 9.3 points versus MD-correlation graphs while needing orders-of-magnitude less compute.
4. The same trick boosts RNA tasks (chemical-modification and binding-site prediction) even though structure predictors are less accurate for RNA, hinting that uncertainty-encoded distances generalise beyond proteins.
5. When plugged into ThermoMPNN for ∆∆G prediction, distogram edge features lift R² from 0.518 → 0.577 and Spearman ρ from 0.726 → 0.749 on the mega-scale stability dataset, again without any MD.
6. Overlap analysis shows distogram edges capture long-range “fuzziness” around loops whereas MD correlations highlight coordinated helix-sheet motions; the two signals are complementary but distograms are free once the structure predictor has run.
7. Limitations: quality still hinges on MSA richness, compute grows for huge complexes, and pairwise marginals can’t disentangle higher-order cooperativities—yet the route to large-scale dynamic modelling without MD is now open.
💻Code: anonymous.4open.science/r/Di…
📜Paper:biorxiv.org/content/10.64898…
#proteindynamics #AlphaFold #graphNN #drugdiscovery #structuralbiology #bioinformatics
10
91
4,521
FLARE: Fine-grained Learning for Alignment of Spectra-molecule Representations Enhances Metabolite Annotation
1. FLARE pushes metabolite annotation to a new SOTA on MassSpecGym: 43.15 % rank@1 by mass and 22.66 % by formula, beating the previous best by > 63 %.
2. Instead of collapsing a spectrum and molecule into single vectors, FLARE keeps peaks and atoms as individual tokens and computes similarity via bidirectional peak-atom maxima, letting local chemical evidence drive the global score.
3. The model is trained only on spectrum–molecule pairs; no peak-to-fragment labels are given, yet the learned alignments agree with MAGMa-predicted fragments, offering an inspectable map of which peak supports which substructure.
4. Spectral and molecular embeddings self-organize into a chemically coherent space: Pearson r ≈ 0.85 between embedding distance and fingerprint Tanimoto, enabling reliable similarity search even across modalities.
5. Compared with global-contrastive JESTR, FLARE wins more often and by larger rank margins, especially on molecules dissimilar to training data, indicating better generalization from local interactions.
6. In a breast-cancer PDX study FLARE recovered 29 of 74 true standards at rank 1, flagged 54 differential metabolites between metastatic and primary tumors, and proposed plausible substructures for 23 low-confidence yet significant features.
7. An interactive Hugging Face demo lets users upload a spectrum and visualize peak-to-node heat-maps in real time, turning black-box retrieval into a chemically traceable workflow.
💻Code: huggingface.co/spaces/Hassou…
📜Paper: biorxiv.org/content/10.64898…
#metabolomics #massspectrometry #graphNN #contrastivelearning #interpretableAI #cheminformatics

3
21
1,459

9 Oct 2025
"Less is more" HRM[0] is very reminiscent of UT[1] which was released in 2019 (and that we spent a lot of time studying at OpenAI in the reasoning team circa 2020). Uses same dynamic halting approach (ACT). I'm a bit suspicious it does appear in their ablation studies.
Exciting that the idea of depth recurrent neural network is making a come back to the spotlight!
The problem with UTs is that they have little parameter counts which prevented them from scaling well in tasks that required knowledge. Since your layer(s) are repeated the only way to scale the parameters is to make the layers bigger which rapidly becomes slow af. Lample came up with product-key memory to solve that problem back then[2] but we never quite made it work on regular pretraining workloads. It was kind of great on graphNN like tasks like SAT solving or simple game, no surprise it's making a come back with ARCAGI.
One interesting idea that we had but never quite explored seriously, circa 2022 this time, was that the nascent MoE architecture could be the answer to make UT work better on regular pre-training. With a MoE/UT you can scale your # of parameters and your depth of recurrence independently to get the best of both world. To this date I'm still surprised that we don't have more UT/MoE since there are many signs that show that recurrence (in depth) is a strong inductive bias for reasoning...
[0] arxiv.org/pdf/2510.04871v1
[1] arxiv.org/abs/1807.03819
[2] arxiv.org/pdf/1907.05242
5
20
185
12,859

9 May 2025
Machine Learning Meets Pharmacokinetics: A Comparative Analysis of Predictive Models for Plasma Concentration-Time Profiles
1.This study systematically compares five machine learning (ML) approaches for predicting in vivo rat plasma concentration-time (PK) profiles from molecular structure, establishing a new benchmark for computational pharmacokinetics.
2.The standout performer was CMT-PINN, a physics-informed neural network trained directly on concentration-time data, achieving the highest R²-log (0.854), Spearman correlation (0.933), and lowest error metrics among all tested models.
3.PURE-ML, a decision tree model trained on log-transformed concentrations, also performed well, with a lower MAPE-log (22.1%) than CMT-PINN, though with slightly less robust overall profile shape recovery.
4.By contrast, traditional hybrid approaches like NCA-ML and PBPK-ML showed much poorer performance, especially in capturing late-phase PK behavior, indicating limits in models trained on derived parameters.
5.All models were evaluated on the same rigorously curated dataset of 696 compounds and 14,155 plasma concentration points from Sanofi's in vivo studies, ensuring fair comparison under standardized metrics.
6.CMT-PINN and PURE-ML both accurately predicted over 60% of concentration points within a two-fold error margin, while NCA-ML and CMT-ML captured less than 12%, highlighting the gap in modeling strategy efficacy.
7.The authors emphasize that training directly on raw concentration-time data, rather than NCA or compartmental model parameters, leads to markedly improved predictive accuracy—especially valuable for small datasets.
8.While PBPK-ML offers mechanistic interpretability, its prediction quality was highly variable and dependent on in vitro parameter prediction, highlighting the challenge of translating early ADME data into robust PK simulations.
9.Graph convolutional neural networks (GCNNs) were used in NCA-ML and CMT-ML to predict PK parameters from SMILES strings, but their performance suffered from poor parameter identifiability and overfitting risks.
10.The CMT-PINN approach required fewer assumptions, smaller architecture, and no preprocessing of PK parameters, making it both more accurate and computationally efficient than many alternatives.
11.Key PK metrics (AUC, C0, Cmin) were best predicted by CMT-PINN and PURE-ML, showing minimal bias, while NCA-ML and PBPK-ML systematically under- or overestimated these values.
12.This paper highlights the importance of model interpretability, evaluation across the entire PK curve, and standardized comparison frameworks—crucial for transitioning these models into real-world discovery pipelines.
13.Future directions include integrating uncertainty quantification, defining model applicability domains, incorporating non-linear ADME mechanisms, and adopting FAIR data practices to improve transparency and adoption.
14.This comprehensive evaluation sets the stage for an "Applied AI Factory" in drug discovery, where validated ML models can reliably inform early PK predictions, reduce in vivo testing, and accelerate design-make-test cycles.
📜Paper: biorxiv.org/content/10.1101/… #Pharmacokinetics #DrugDiscovery #MachineLearning #PINN #ADME #PKModeling #ComputationalPharmacology #GraphNN #Bioinformatics #AI4Science

1
4
837

28 Apr 2025
An GraphNN Bayesian local learning with SWT data of the price for Reversal estimation is to be considered to increase the stability of the signal
1
3
28

28 Mar 2024
Hace dos semanas, comenzamos nuestro curso para celebrar el día pi: Graph Neural Networks & AlphaFold.
¡Recuerda que si eres nuestro alumno, tienes acceso gratuito a este y otros recursos de ciencia de datos!
#GraphNN #AlphaFold #Proteinas

1
118

21 Feb 2024
Excited to kick off our first @pydataberlin Meetup of the year! 🎉 Dr. Martin Hanik is diving into Exploiting Data Structure and Geometry with a Diffusion-based Graph Neural Network. Huge thanks to @GetYourGuide for hosting us again! #PyData #DataScience #MachineLearning #GraphNN

1
2
204
19 Feb 2024
¡Estamos muy emocionados por iniciar nuestras terceras lecciones semestrales!
En esta ocasión, presentaremos Graph Neural Networks y AlphaFold de la mano de Álvaro de Obeso.
¡Estas lecciones son gratuitas y dirigidas a nuestros estudiantes!
#Lecciones #AlphaFold #GraphNN

2
71

3 Oct 2023
Unlocking the secrets of graph neural networks with Prof. Michael Bronstein (@mmbronstein) 🚀. Symmetry, permutation & power in the spotlight! 🔍
@GoogleDeepMind @UniofOxford
#AIResearch #GraphNN #TechInsights #MachineLearning #DeepLearning #AI #OxML #GlobalGoalsAi #AI4GG
1
7
479

13 Jun 2023
分子LLMの動向をざっくりと把握。SMILES文に埋め込まれた分子の特徴(三次元構造とか)の表現学習がタンパク質LLMに比べるとあまり上手くいってないという感じなのかな? VAEはいまいちだったとしても非SMILESベースの方法(従来型のフィンガープリントとか、graphNNとか)と比べてどうなんだろ?
3
8
1,073

16 May 2023
(2/n) We present the use of graphNN in autoencoders, showcasing great efficiency in various problems, also bifurcation, and low data regimes.
So thankful for this collaboration with Dr. Federico Pichi and Prof. Jan S Hesthaven at @EPFL
1
2
143

20 Jan 2023
Saved this Tweet to your Notion database.
Tags: [Graphnn]
38

4 Jan 2023
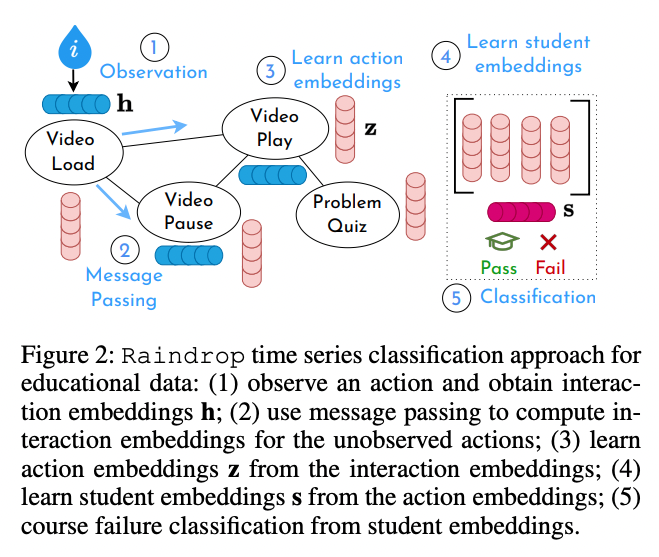
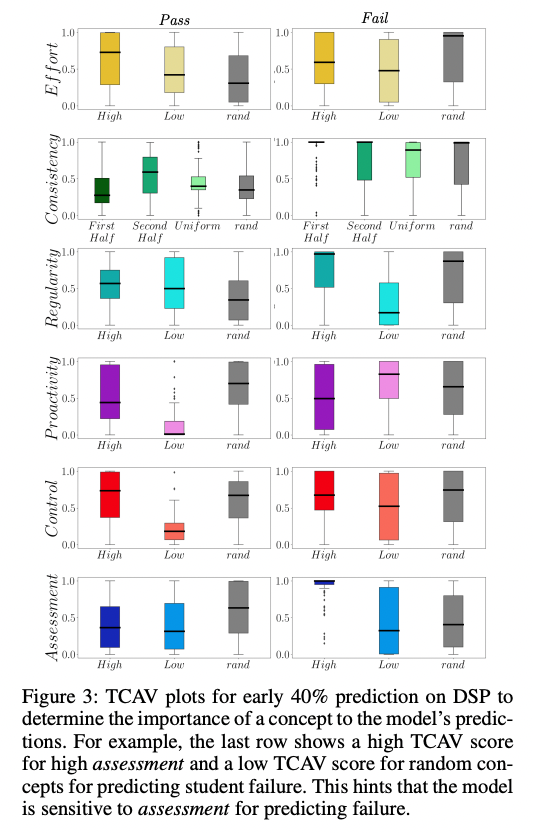
See you at AAAI this year! We present RIPPLE, an interpretable graphNN prediction pipeline using raw time series student clickstreams. Comparable accuracy to SoTA models✨with no feature extraction✨ plus interpretability for personalized interventions. arxiv.org/abs/2212.01133

2
1
21
2,147

28 Dec 2022
PhD studentship "Leveraging novel knowledge base and Graph Representation Learning for Mobile Mental Health Social Networks", #NLP #GraphNN #SNA
10 Oct 2022

New Fully funded #PhD #studentship.
I am looking for an enthusiastic student to work on this exciting project #NaturalLanguageProcessing #NLP, #MachineLearning #AI #AffectiveComputing & #SocialNetoworkAnalysis #EmotionRecognition #GraphNN #SNA
#Nottingham lnkd.in/eVVwjxnv
2
175

10 Oct 2022
New Fully funded #PhD #studentship.
I am looking for an enthusiastic student to work on this exciting project #NaturalLanguageProcessing #NLP, #MachineLearning #AI #AffectiveComputing & #SocialNetoworkAnalysis #EmotionRecognition #GraphNN #SNA
#Nottingham lnkd.in/eVVwjxnv
7
9

29 Sep 2022
pubs.acs.org/doi/10.1021/acs…
溶解度予測を、分子記述子NN, GraphNN, LSTM with SMILES, SchNETで比較。学習データは16000 個。分子記述子が最も良い精度。多くデータを集めると、アーキテクチャはあまり関係がなくなってくるとのこと。。。
#マテリアルズ・インフォマティクス #MI_Papers
1
28

21 Sep 2022
It was a full house at Ekaterina Sirzitdinova's #GraphNN lab! For those who didn't make it - check out Accelerating GNNs with PyTorch Geometric and GPUs. It'll also be on demand after the broadcast for GTC registrants.
Watch Sept 22, 4pm CEST at #GTC22 > nvda.ws/3eWxuzp

1
4

15 May 2022
ん~~~~Transformerが言語処理以外でもこんなにメインストリームになるとは思わなかったな。GraphNNもモノになるとは思ってなかったし、機械学習とかいうの全然分かんないな。
4

7 Mar 2022
Advances of ML Approaches for Financial Decision Making & Time Series Analysis – Track spotlight ☄️
How can #Finance fully use #ML? (#DeepLearning, #RL, #NLP & #GraphNN)
Join us on March 28 at #AMLDEPFL22 – organized by @ETH_en & @nyuniversity
More info: buff.ly/3MHcysT

1
2