
🚀 Key Read in #AI Research
AutoML just got faster & more transparent ⚡Meet SIBILA: an AutoML framework built for HPC platforms that doesn't sacrifice interpretability. Competitive accuracy regulatory-ready models. #AutoML #InterpretableAI #HPC
Read: doi.org/10.3390/ai5040116

18

Jun 6
🎓 Fully Funded PhD in 𝗜𝗻𝘁𝗲𝗿𝗽𝗿𝗲𝘁𝗮𝗯𝗹𝗲 𝗔𝗜 𝗳𝗼𝗿 𝗖𝗼𝘂𝗽𝗹𝗲𝗱 𝗠𝘂𝗹𝘁𝗶𝗽𝗵𝘆𝘀𝗶𝗰𝘀 𝗼𝗳 𝗚𝗲𝗼𝗺𝗮𝘁𝗲𝗿𝗶𝗮𝗹𝘀 (Switzerland 🇨🇭)
🏆 EPFL— Ranked #22 globally in the QS World University Rankings, is one of Europe's premier engineering and technology universities.
💶 Fully funded 4️⃣ year PhD with a competitive doctoral salary and excellent benefits
✅ Passionate about #ArtificialIntelligence #ComputationalMechanics #Geomaterials 🤖⛰️⚙️
✅ Highly recommend this interdisciplinary #fullyfunded #PhDPositionwithin the Data-Driven Mechanics Laboratory @EPFL_en 🇨🇭
📌 This #phdproject focuses on developing interpretable AI and physics-informed machine learning methods to discover governing equations and constitutive laws for complex multiphysics processes in geomaterials, with applications in geophysics, energy, and sustainability.
You’ll work on:
🔷 Developing next-generation data-driven frameworks for scientific discovery in geomaterials
🔷 Combining computational mechanics with interpretable and thermodynamics-informed machine learning
🔷 Discovering governing equations and constitutive relations from rich experimental and micromechanical datasets
🔷 Building multiscale models spanning discrete, mesoscale, and continuum mechanics
🔷 Applying neurosymbolic AI approaches to uncover physically meaningful relationships across scales
🔷 Collaborating internationally with leading experimental research groups in geomechanics and materials science.
🌍 Contribute to advancing sustainable energy systems, geophysics, and earth sciences by creating interpretable AI tools that reveal the fundamental physical mechanisms governing complex geomaterial behaviour.
✅ Work with Prof. @kon_karapiperis , and the Data-Driven Mechanics Laboratory @EPFL_en
⏰ 𝗗𝗲𝗮𝗱𝗹𝗶𝗻𝗲: 𝟭𝟱𝘁𝗵 𝗝𝘂𝗹𝘆, 𝟮𝟬𝟮𝟲
👉 Full details & apply here:
🔗phdscanner.com/opportunities…
📩 Want more like this?
➕ Follow @PhDScanner and join WhatsApp for updates:
whatsapp.com/channel/0029Vb5…
🌐 Visit: phdscanner.com
#fullyfundedPhD #PhDposition #EPFL #Switzerland #ArtificialIntelligence #ScientificMachineLearning #ComputationalMechanics #Geomaterials #InterpretableAI #PhysicsInformedAI #Geophysics #ComputationalScience #ResearchOpportunity
@phdhardtalk
♻️ Share with someone applying this cycle

ALT https://www.phdscanner.com/opportunities/phd-vacancies-epfl-switzerland-phd-in-interpretable-ai-for-coupled-multiphysics-of-geomaterials-7177a261-0c47-4903-825a-e908e80453f9
1
6
486

May 24
The third session of BIOINFOCONGRESS VIII is now available.
Assoc. Prof. Erdal Toprak’s talk titled
“Toward Impeding and Even Reversing Antibiotic Resistance with Interpretable Artificial Intelligence Models”
along with the Q&A session is now on the Bioinforange YouTube channel.
Bring your scientific curiosity and your headphones.
YouTube: Bioinforange
#bioinforange #bioinfocongressviii #antibioticresistance #interpretableai #systemsbiology
1
5
151
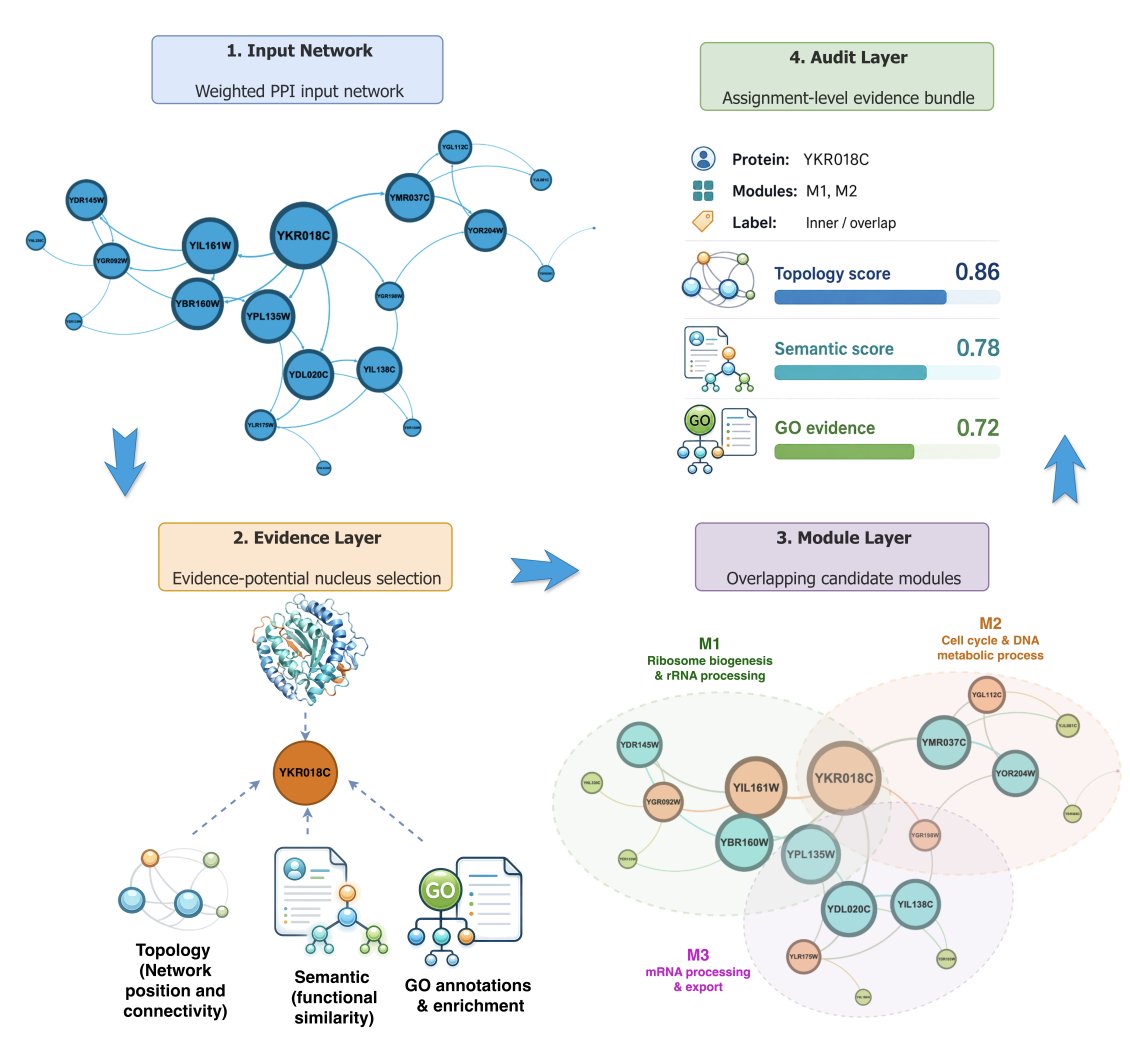
ECHO-PPI: Trustworthy AI for Evidence-Bundled Detection of Overlapping Protein Modules in Protein–Protein Interaction Networks
1. ECHO-PPI is an evidence-bundled framework for overlapping module detection in PPI networks that explicitly targets curator-facing interpretability: every protein–module assignment is exported with an auditable bundle of topology, semantic, and Gene Ontology (GO) evidence, plus a hierarchical confidence label (core/inner/outer/uncertain).
2. The key design shift is from “module lists” to “assignment-level decision support”: instead of only returning overlapping communities, ECHO-PPI records why a specific protein is placed into a specific module, and whether that membership should be treated as strong (core) or boundary/triage (inner/outer/uncertain).
3. The workflow integrates three evidence channels: (i) weighted PPI topology, (ii) semantic protein profiles (TF–IDF SVD embeddings of text/GO labels; Sentence-BERT is optional but not used in reported benchmarks), and (iii) GO evidence via module-specific GO TF–IDF “functional signatures,” while treating missing GO as missing evidence rather than biological absence.
4. For overlap-aware membership scoring, ECHO-PPI combines a boundary-sensitive topology metric (permanence) with functional dependency (alignment of a protein’s GO terms to a module’s GO TF–IDF signature) using a transparent mixture score M(p, C) = α Perm(p, C) (1−α) fd(p, C). Overlaps are added only if they improve the best existing assignment by a margin (gain threshold), with a conservative transfer rule for likely misplacements.
5. Candidate generation is broadened beyond a single clustering output: starting from MCL modules, ECHO-PPI adds nucleus-centered ego neighborhoods (1–2 hops), greedy topology–semantic expansions, semantic kNN sets that pass a graph-support filter, and hybrid unions when candidate Jaccard overlap is high; candidates are then scored with penalties for uncertainty and fragmentation.
6. A distinctive component is the deterministic “evidence-potential nucleus” score (BH), inspired by gravity-based representative selection, to prioritize high-support local nuclei using weighted degree, clustering coefficient, k-core, semantic neighborhood coherence, GO richness, and an annotation-sparsity penalty. Importantly, nuclei guide candidate construction and ranking rather than defining the final partition alone.
7. ECHO-PPI adds “recall-safe supplementation” to avoid the common failure mode of naive boundary expansion: it limits growth (≤15% relative size increase, at most two added proteins per module) and requires gated evidence gain, so expansions remain conservative and reviewable.
8. Confidence labeling is hierarchical and evidence-based: core requires both topology and semantic support above thresholds; inner/outer require weaker support; uncertain captures boundary cases. The paper emphasizes these labels as triage metadata (especially core vs non-core), not as calibrated probabilities, and reports how label distributions can shift with preprocessing and embedding normalization.
9. Benchmarking on yeast (Gavin socioaffinity network; plus a Krogan 2006 BioGRID-derived transfer benchmark) shows ECHO-PPI largely preserves the predictive behavior of the MCL overlap seed rather than outperforming the strongest baseline (ClusterONE). On Gavin full-gold, ClusterONE leads (F1 0.270) while ECHO-PPI matches MCL-scale performance (F1 0.162) but uniquely delivers complete required-field evidence-bundle coverage (1.00 vs 0.00 for baselines).
10. The paper’s central claim is therefore complementary to pure F1 optimization: ECHO-PPI makes overlapping module predictions inspectable, confidence-aware, and reproducible. Case studies (e.g., YKR018C, YIL161W) illustrate multi-membership outputs where semantic evidence can dominate topology in some assignments, explicitly signaling “hypotheses for manual review” rather than silently promoting all overlaps to equally strong complex memberships.
💻Code: github.com/MehrdadJalali-AI/…
📜Paper: arxiv.org/abs/2605.21216
#computationalbiology #bioinformatics #PPI #proteininteractions #networkscience #communitydetection #interpretableAI #trustworthyAI #GeneOntology #reproducibility
3
14
1,234

May 22
Today I’m sharing a new scientific milestone:
“Congruity as a Candidate Structural Invariance Class: Interpretable Cross-Domain Symbolic Emergence, Falsification, and Transfer Evaluation”
Preprint: doi.org/10.5281/zenodo.20349…
Public reproducibility repository: github.com/andrearomeo74-clo…
This work does not claim a final universal equation.
Instead, it asks a narrower and more testable scientific question:
Do viable heterogeneous systems repeatedly exhibit interpretable proportional structural patterns under interacting burdens?
To explore this, the study evaluates reproducible benchmarks across multiple domains:
• breast cancer morphology classification
• external unseen diabetes clinical validation
• synthetic ecological collapse dynamics
• symbolic structural discovery
• permutation falsification
• proxy robustness analysis
• adaptive leave-one-domain-out transfer
• fixed-sign transfer stress testing
Core observations:
• interpretable proportional structures repeatedly emerge
• symbolic search converges toward multiplicative burden forms
• randomized falsification materially degrades performance
• transfer remains partially preserved across domains
• some assumptions fail under stress, which is scientifically informative
This is important because the goal is not to defend a theory at all costs.
The goal is to determine whether Congruity represents a genuine structural invariance family, or a domain-specific artifact.
Everything public here is intentionally reproducible.
Independent verification, critique, replication, and failure analysis are welcome.
Science progresses through exposure, not insulation.
#Science #ComplexSystems #AI #MachineLearning #SystemsBiology #InterpretableAI #SymbolicAI #Reproducibility #TransferLearning #SystemsScience #ComputationalScience #OpenScience

2
4
466

🚨 Medical AI Research Alert! 🚨
How can we make AI disease screening both highly performant AND truly interpretable, mirroring how clinicians think?
@HongKongPolyU presents 𝗘𝘃𝗶𝗦𝗰𝗿𝗲𝗲𝗻: 𝗮𝗻 𝗲𝘃𝗶𝗱𝗲𝗻𝘁𝗶𝗮𝗹 𝗿𝗲𝗮𝘀𝗼𝗻𝗶𝗻𝗴 𝗳𝗿𝗮𝗺𝗲𝘄𝗼𝗿𝗸 𝗹𝗲𝘃𝗲𝗿𝗮𝗴𝗶𝗻𝗴 𝗿𝗲𝗴𝗶𝗼𝗻-𝗹𝗲𝘃𝗲𝗹 𝗲𝘃𝗶𝗱𝗲𝗻𝗰𝗲 𝗳𝗿𝗼𝗺 𝗵𝗶𝘀𝘁𝗼𝗿𝗶𝗰𝗮𝗹 𝗰𝗮𝘀𝗲𝘀.
By Chenyu Lian, @HongYuZhou14, Jing Qin.
Now you can watch and listen to the latest Medical AI papers daily on our YouTube and Spotify channels!
YouTube Explainer: youtu.be/FcDZturRXvc
YouTube Shorts: youtube.com/shorts/FQYOdW-wa…
Spotify: open.spotify.com/show/4edRuS…
Here's why it's exciting: 👇🧵 1/9
#MedicalAI #Healthcare #DiseaseScreening #InterpretableAI
[1/9]

3
4
13
919
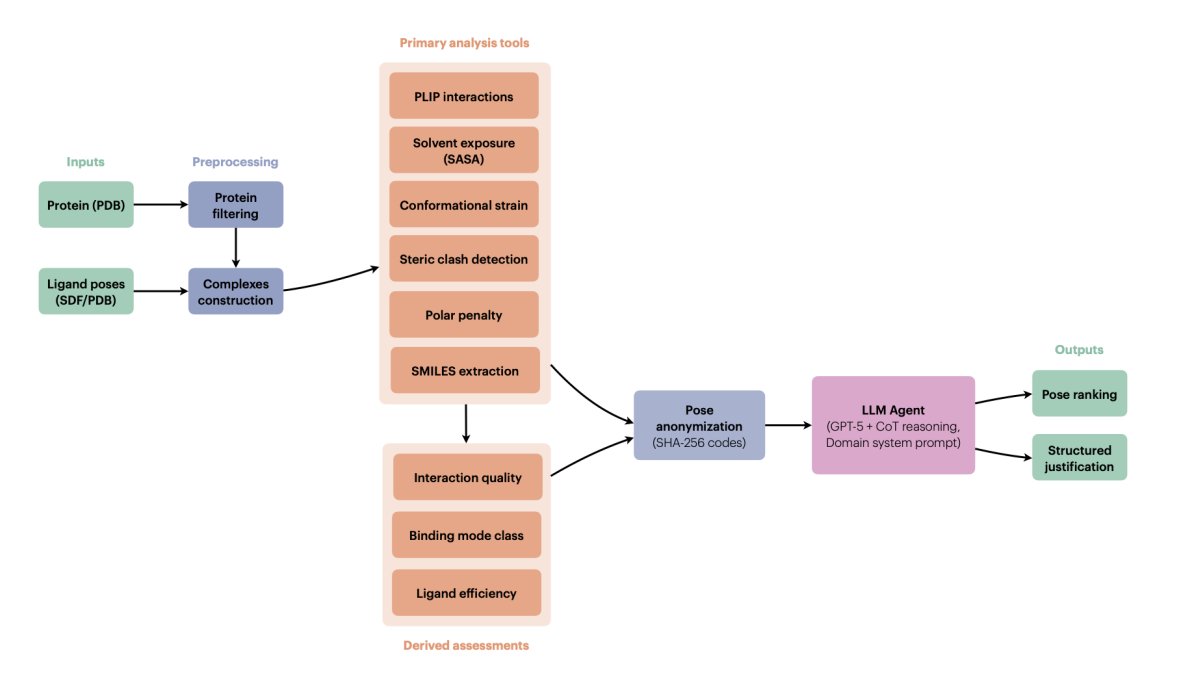
AgenticPosesRanker: An agentic AI framework for physically grounded ranking of protein–ligand docking poses
1. The paper proposes a tool-augmented LLM “pose judge” that ranks docking poses by explicit multi-criteria reasoning over physically interpretable metrics, rather than learning or fitting a new scoring function.
2. AgenticPosesRanker combines 6 deterministic analysis tools (PLIP interaction fingerprinting, SASA/burial, MMFF94-based conformational strain, steric clash detection, unsatisfied buried polar atom penalty, and SMILES identity checks) plus 3 derived assessments (interaction quality scoring, binding-mode classification, ligand efficiency).
3. Key design choice: keep each physicochemical observable separate and interpretable, then let the LLM integrate conflicting evidence (e.g., strong interactions vs. high strain, deep burial vs. polar desolvation risk) with a structured decision rubric instead of fixed weights.
4. The agent uses GPT-5 with chain-of-thought enabled and a long domain-specific system prompt that encodes tool documentation, thresholds, and a hierarchical ranking policy (e.g., binding mode and interaction quality prioritized; strain/clashes/polar penalties as discriminators; “surface binders should never rank first”).
5. To reduce name-based leakage (e.g., pose_01 implying docking-score order), poses are anonymized via deterministic SHA-256-derived 8-character base-36 codes; tool outputs are sanitized to remove original identifiers before being passed to the LLM, then de-anonymized after inference.
6. Benchmark: 10 protein–ligand systems, 162 poses, curated to be balanced “by construction” between cases where Smina ranks a near-native pose correctly and cases where Smina fails. Ground truth is RMSD to crystallographic pose.
7. Main result: 50.0% best-pose accuracy on the 10-system benchmark, matching the design-fixed Smina baseline (50.0%) and far above a uniformly random baseline (7.7%; p < 0.001, one-sided exact binomial test). The aggregate hides symmetry: 80% retention of Smina successes (4/5) but only 20% recovery of Smina failures (1/5).
8. A notable methodological contribution is “decision attribution” analysis: the paper compares the agent’s self-reported tool weighting against objective metric separations for the selected pose, finding strong alignment (median Spearman ρ = 0.83) across both correct and incorrect outcomes—suggesting the limiting factor is tool-suite coverage, not inconsistent reasoning.
9. The work emphasizes interpretability and auditability: instead of an opaque scalar score, the output includes metric-by-metric justification and structured comparative analysis, positioning the agent as an interpretable curation layer for late-stage pose refinement rather than a guaranteed accuracy upgrade over classical scoring.
📜Paper: arxiv.org/abs/2605.03707
#computationalbiology #computationalchemistry #moleculardocking #structurebaseddrugdesign #LLM #agenticAI #interpretableAI #drugdiscovery #proteinligand #cheminformatics
2
1,045
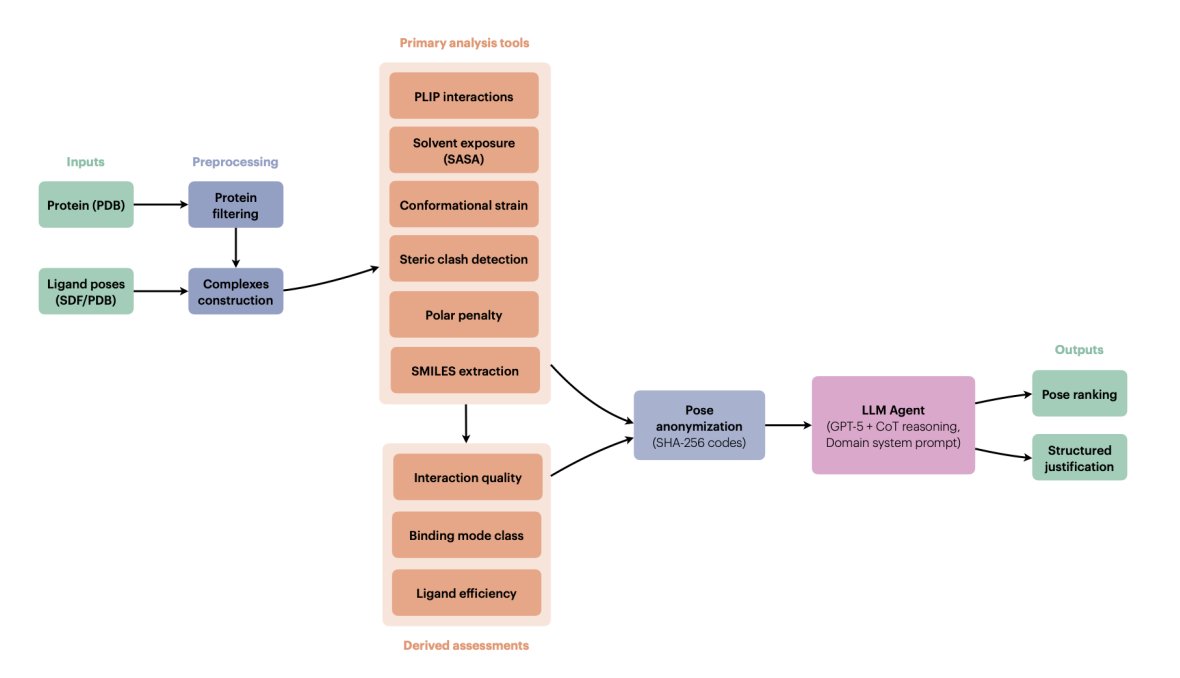
AgenticPosesRanker: An agentic AI framework for physically grounded ranking of protein–ligand docking poses
1. The paper proposes a tool-augmented LLM “pose judge” that ranks docking poses by explicit multi-criteria reasoning over physically interpretable metrics, rather than learning or fitting a new scoring function.
2. AgenticPosesRanker combines 6 deterministic analysis tools (PLIP interaction fingerprinting, SASA/burial, MMFF94-based conformational strain, steric clash detection, unsatisfied buried polar atom penalty, and SMILES identity checks) plus 3 derived assessments (interaction quality scoring, binding-mode classification, ligand efficiency).
3. Key design choice: keep each physicochemical observable separate and interpretable, then let the LLM integrate conflicting evidence (e.g., strong interactions vs. high strain, deep burial vs. polar desolvation risk) with a structured decision rubric instead of fixed weights.
4. The agent uses GPT-5 with chain-of-thought enabled and a long domain-specific system prompt that encodes tool documentation, thresholds, and a hierarchical ranking policy (e.g., binding mode and interaction quality prioritized; strain/clashes/polar penalties as discriminators; “surface binders should never rank first”).
5. To reduce name-based leakage (e.g., pose_01 implying docking-score order), poses are anonymized via deterministic SHA-256-derived 8-character base-36 codes; tool outputs are sanitized to remove original identifiers before being passed to the LLM, then de-anonymized after inference.
6. Benchmark: 10 protein–ligand systems, 162 poses, curated to be balanced “by construction” between cases where Smina ranks a near-native pose correctly and cases where Smina fails. Ground truth is RMSD to crystallographic pose.
7. Main result: 50.0% best-pose accuracy on the 10-system benchmark, matching the design-fixed Smina baseline (50.0%) and far above a uniformly random baseline (7.7%; p < 0.001, one-sided exact binomial test). The aggregate hides symmetry: 80% retention of Smina successes (4/5) but only 20% recovery of Smina failures (1/5).
8. A notable methodological contribution is “decision attribution” analysis: the paper compares the agent’s self-reported tool weighting against objective metric separations for the selected pose, finding strong alignment (median Spearman ρ = 0.83) across both correct and incorrect outcomes—suggesting the limiting factor is tool-suite coverage, not inconsistent reasoning.
9. The work emphasizes interpretability and auditability: instead of an opaque scalar score, the output includes metric-by-metric justification and structured comparative analysis, positioning the agent as an interpretable curation layer for late-stage pose refinement rather than a guaranteed accuracy upgrade over classical scoring.
📜Paper: arxiv.org/abs/2605.03707
#computationalbiology #computationalchemistry #moleculardocking #structurebaseddrugdesign #LLM #agenticAI #interpretableAI #drugdiscovery #proteinligand #cheminformatics
2
3
863
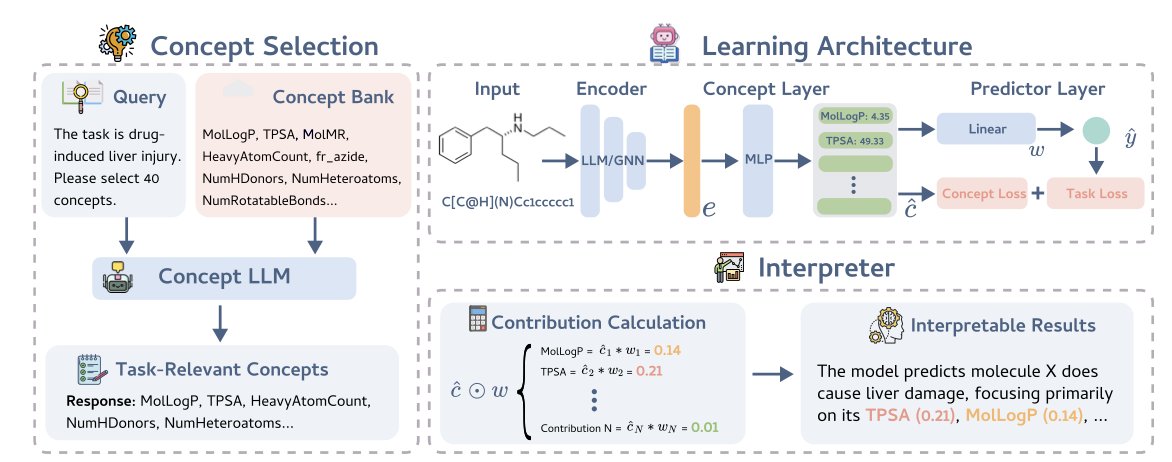
GlassMol: Interpretable Molecular Property Prediction with Concept Bottleneck Models
1. The paper introduces GlassMol, a model-agnostic Concept Bottleneck Model (CBM) framework that challenges the long-assumed trade-off between interpretability and performance in molecular property prediction.
2. Unlike black-box Graph Neural Networks and Large Language Models, GlassMol projects molecular embeddings onto human-interpretable chemical concepts before making predictions, ensuring faithful and transparent reasoning.
3. The framework addresses three critical gaps in applying CBMs to chemistry: the Relevance Gap through LLM-guided concept selection, the Annotation Gap via automated RDKit descriptor computation, and the Capacity Gap through empirical demonstration that interpretability need not sacrifice accuracy.
4. GlassMol employs a two-stage curation pipeline where RDKit generates 200 physicochemical descriptors as ground truth, and GPT-4 selects the most task-relevant subset, eliminating manual annotation burden.
5. Experiments across thirteen Therapeutics Data Commons benchmarks show that GlassMol consistently matches or exceeds black-box baselines, with particularly strong gains in toxicity prediction tasks where understanding structural alerts is crucial.
6. The architecture supports both GNN and LLM backbones, with linear prediction layers enabling exact decomposition of contributions from individual chemical concepts such as LogP, TPSA, and heavy atom counts.
7. Case studies demonstrate that GlassMol's concept attributions align with established structural importance methods like TopoPool, correctly identifying known metabolic liabilities and toxicophores in molecules like Famciclovir and Mitomycin C.
8. Ablation studies reveal that domain-specific pre-trained models outperform general-purpose LLMs for molecular tasks, and that open-source Llama-3-70B can serve as a viable alternative to GPT-4 for concept selection.
9. The framework shows remarkable robustness to noisy concept labels, suggesting it learns meaningful chemical patterns rather than memorizing exact descriptor values.
💻Code: github.com/rivera-lanasm/Gla…
📜Paper: arxiv.org/abs/2603.01274
#MachineLearning #DrugDiscovery #MolecularPropertyPrediction #InterpretableAI #ConceptBottleneckModels #Cheminformatics #GNN #LLM #RDKit #TherapeuticsDataCommons
1
2
20
1,713
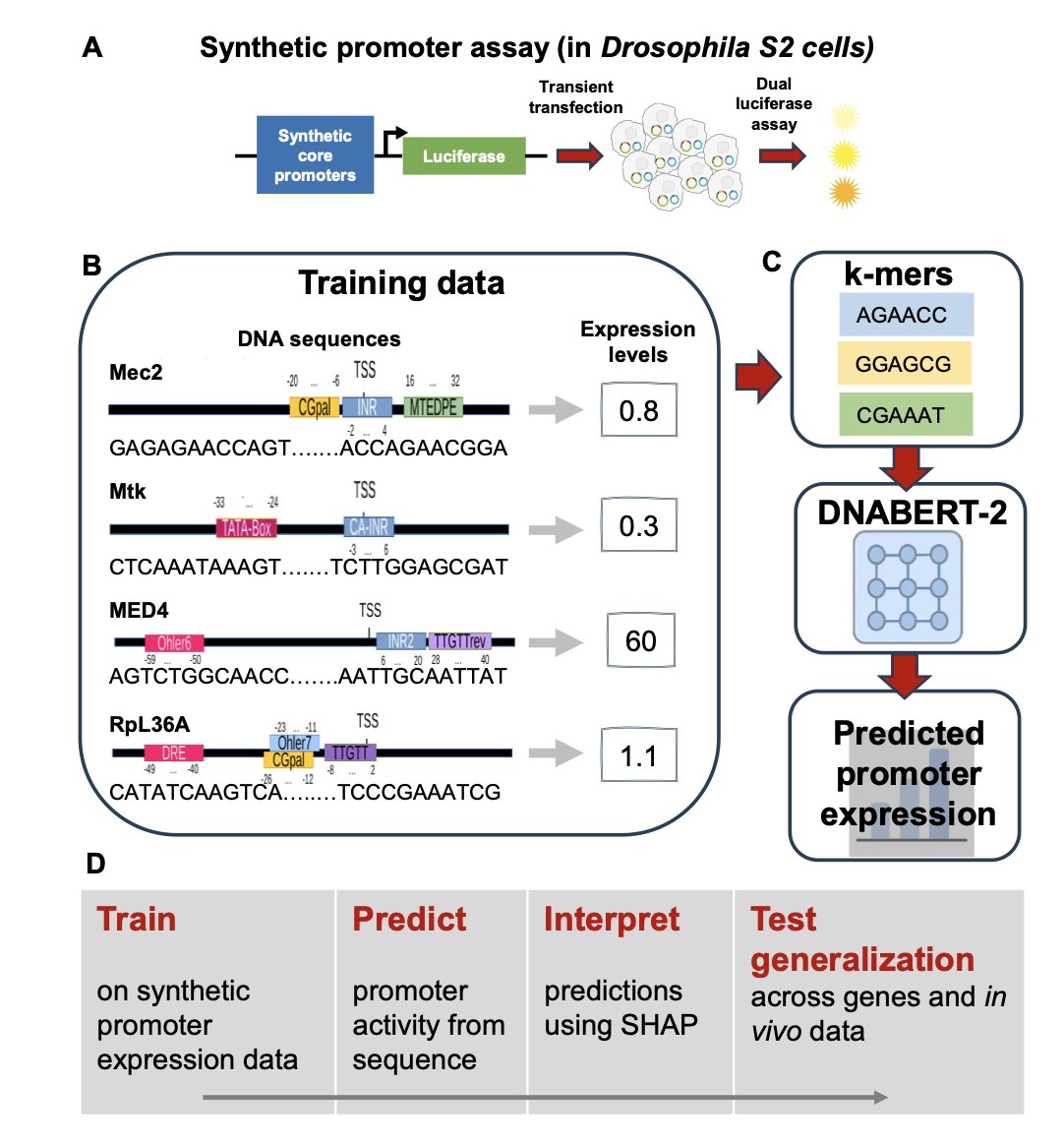
Decoding Promoter Activity from DNA Sequence using Pre-trained Language Models
1 A new study demonstrates that DNABERT-2, a transformer-based DNA language model, can predict Drosophila core promoter activity directly from sequence with remarkable accuracy (R² ≈ 0.91), challenging the notion that promoter regulation requires predefined motif annotations.
2 The research reveals that fine-tuning a 117-million-parameter pre-trained model on just ~700 synthetic promoters with ~2,600 measurements captures quantitative expression patterns without explicit feature engineering, opening new avenues for interpretable deep learning in regulatory genomics.
3 Using SHAP analysis for model interpretability, the authors show that the model autonomously learns biologically meaningful features corresponding to canonical core promoter elements including INR, TATA-box, DPE/MTEDPE, and Ohler motifs, validating its biological relevance.
4 The study systematically tests how biological context modulates promoter activity by integrating ecdysone hormonal signaling and flanking nucleosomal sequences, demonstrating that the unified framework preserves strong predictive performance while capturing known regulatory effects.
5 Gene-wise cross-validation reveals robust generalization across most developmental and constitutive promoters, though motif-less promoters show more variable performance, suggesting these depend more on chromatin state and higher-order regulatory mechanisms.
6 When applied to independent in vivo embryo data without retraining, the model achieves moderate generalization (Pearson r ≈ 0.63), indicating that core promoter sequence explains substantial but incomplete components of transcriptional regulation in complex biological contexts.
7 The work establishes a quantitative framework for rational promoter design and synthetic biology applications, combining accurate prediction with clear interpretability through attribution methods that identify regulatory sequence features directly from DNA.
📜Paper: biorxiv.org/content/10.64898…
#PromoterBiology #DNABERT #DeepLearning #RegulatoryGenomics #Drosophila #SyntheticBiology #TransformerModels #InterpretableAI #Genomics #Bioinformatics
4
11
1,421

📢 #CallforPapers in Electronics
Causal and Structured Representations for Trustworthy and #InterpretableAI
Guest Editor: Guangyi Chen from @CarnegieMellon
Deadline: 15 October 2026
👉 Learn more and submit your manuscript:
mdpi.com/journal/electronics…
#TrustworthyAI #ExplainableAI

1
2
49
FLARE: Fine-grained Learning for Alignment of Spectra-molecule Representations Enhances Metabolite Annotation
1. FLARE pushes metabolite annotation to a new SOTA on MassSpecGym: 43.15 % rank@1 by mass and 22.66 % by formula, beating the previous best by > 63 %.
2. Instead of collapsing a spectrum and molecule into single vectors, FLARE keeps peaks and atoms as individual tokens and computes similarity via bidirectional peak-atom maxima, letting local chemical evidence drive the global score.
3. The model is trained only on spectrum–molecule pairs; no peak-to-fragment labels are given, yet the learned alignments agree with MAGMa-predicted fragments, offering an inspectable map of which peak supports which substructure.
4. Spectral and molecular embeddings self-organize into a chemically coherent space: Pearson r ≈ 0.85 between embedding distance and fingerprint Tanimoto, enabling reliable similarity search even across modalities.
5. Compared with global-contrastive JESTR, FLARE wins more often and by larger rank margins, especially on molecules dissimilar to training data, indicating better generalization from local interactions.
6. In a breast-cancer PDX study FLARE recovered 29 of 74 true standards at rank 1, flagged 54 differential metabolites between metastatic and primary tumors, and proposed plausible substructures for 23 low-confidence yet significant features.
7. An interactive Hugging Face demo lets users upload a spectrum and visualize peak-to-node heat-maps in real time, turning black-box retrieval into a chemically traceable workflow.
💻Code: huggingface.co/spaces/Hassou…
📜Paper: biorxiv.org/content/10.64898…
#metabolomics #massspectrometry #graphNN #contrastivelearning #interpretableAI #cheminformatics

3
21
1,459

Self-driving vehicles can be a model for building trust in physical AI. Nuro Co-founder & Co-CEO @dfergusonnz explores a framework for facilitating open dialogues in an age where technology is learning to speak for itself. Read the Forum Stories article here: weforum.org/stories/2026/01/…
#WEF26 #SelfDriving #AutonomousVehicles #PhysicalAI #InterpretableAI
3
5
28
1,935
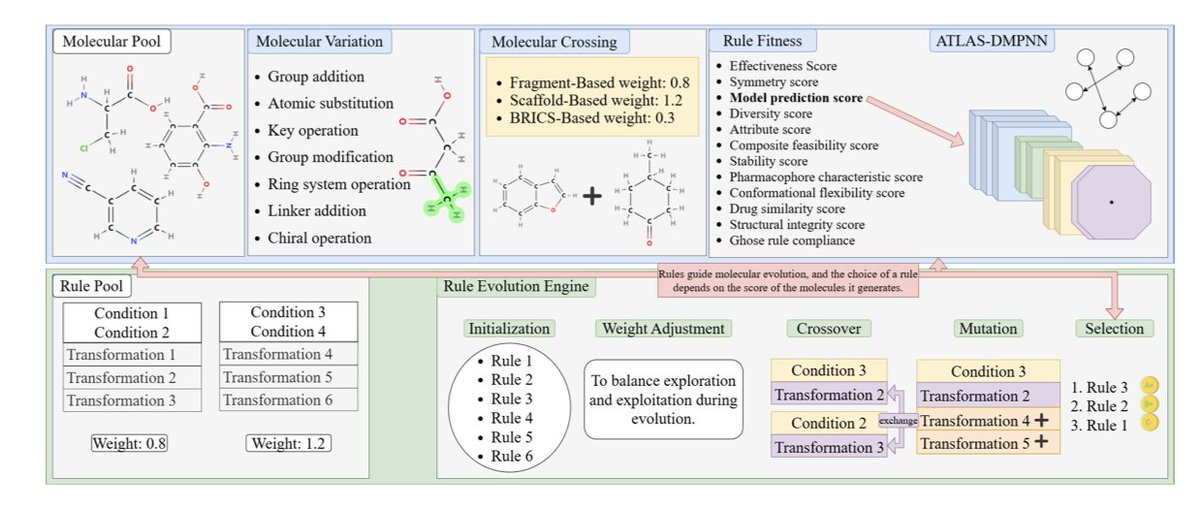
Multi-Constrained Evolutionary Molecular Design Framework: An Interpretable Drug Design Method Combining Rule-Based Evolution and Molecular Crossover
1. The study introduces MCEMOL, a novel framework for molecular optimization that integrates rule-based evolution with molecular crossover, offering an interpretable and efficient approach to drug design. Unlike traditional deep learning methods, MCEMOL requires minimal computational resources and evolves from a small number of starting molecules.
2. MCEMOL employs a dual-layer evolutionary strategy, optimizing transformation rules at the rule level while applying crossover and mutation to molecular structures. This dual approach ensures high molecular validity, diversity, and drug-likeness compliance, achieving 100% validity in generated molecules.
3. The framework incorporates a comprehensive chemical constraint system and message-passing neural networks to guide molecular optimization. It balances interpretability with generation performance, providing transparent design pathways through its evolutionary mechanism.
4. MCEMOL demonstrates strong performance in symmetry constraints, pharmacophore optimization, and stereochemical integrity. It delivers dual outputs: interpretable transformation rules that researchers can understand and trust, and a high-quality molecular library ready for practical applications.
5. Experimental results show that MCEMOL outperforms existing methods in terms of novelty, diversity, and drug-likeness while maintaining high interpretability. The framework is computationally efficient, making it accessible for resource-limited research settings.
📜Paper: arxiv.org/abs/2601.10110v1
#MolecularDesign #DrugDiscovery #EvolutionaryAlgorithm #InterpretableAI #ComputationalBiology
2
16
1,333

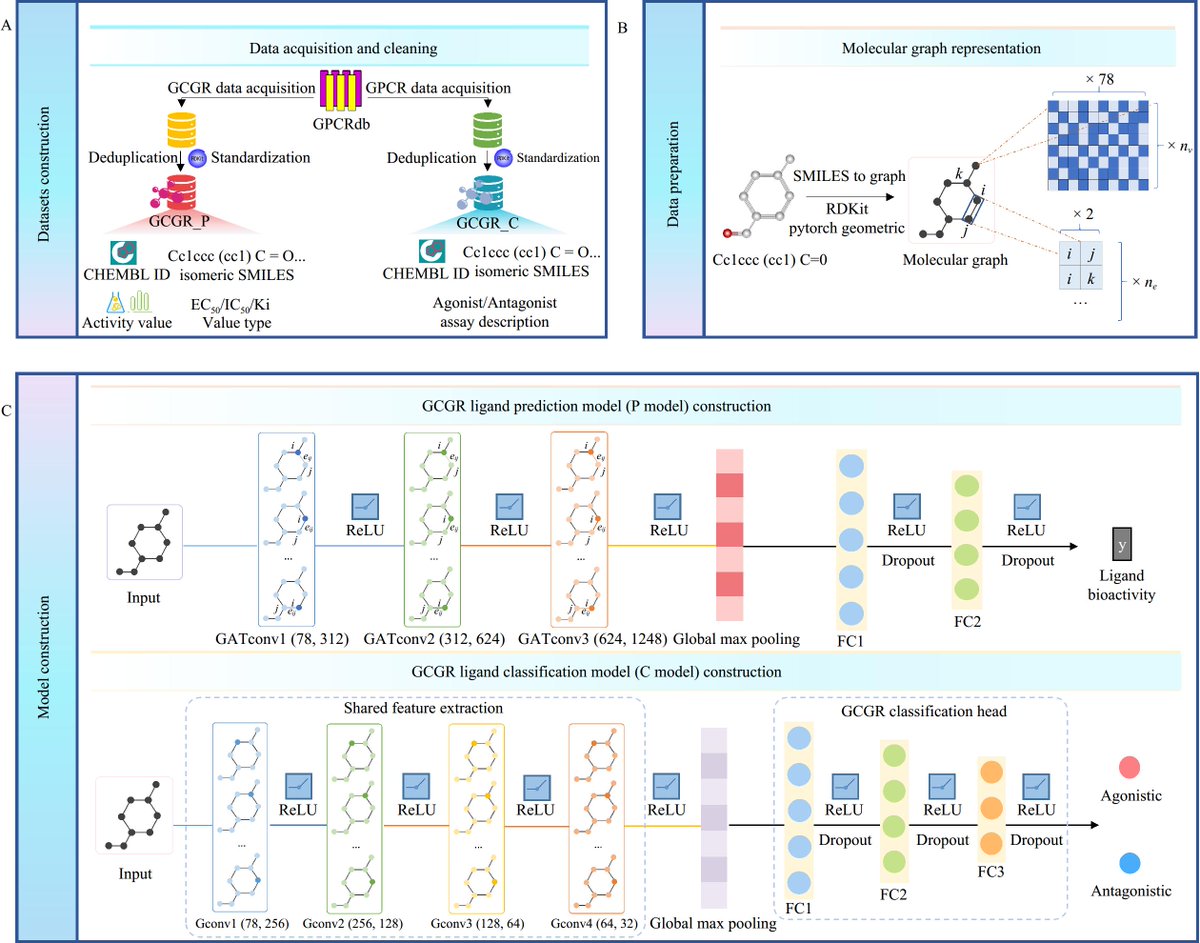
🚀 DeepGCGR: Interpretable AI for GCGR agonists!
Combines GCN attention layers to predict bioactivity & functional effects, accelerating T2DM drug discovery.
🔗 Dive into the full study:
sciencedirect.com/science/ar…
🔗Doi: doi.org/10.1016/S1875-5364(2…
#AI #DrugDiscovery #DeepLearning #GCN #InterpretableAI #T2DM #CJNM
3
5
48

20 Dec 2025
📅 #IndoML2025 | Day 2 – Session 2
⏰ 2:00–2:45 PM
🎙️ Utkarsh Mall (MBZUAI)
🧠 Interpretable Programs for Visual Discovery in Science
📍 BITS Pilani Hyderabad Campus
#InterpretableAI #IndoML
2
86

9 Dec 2025
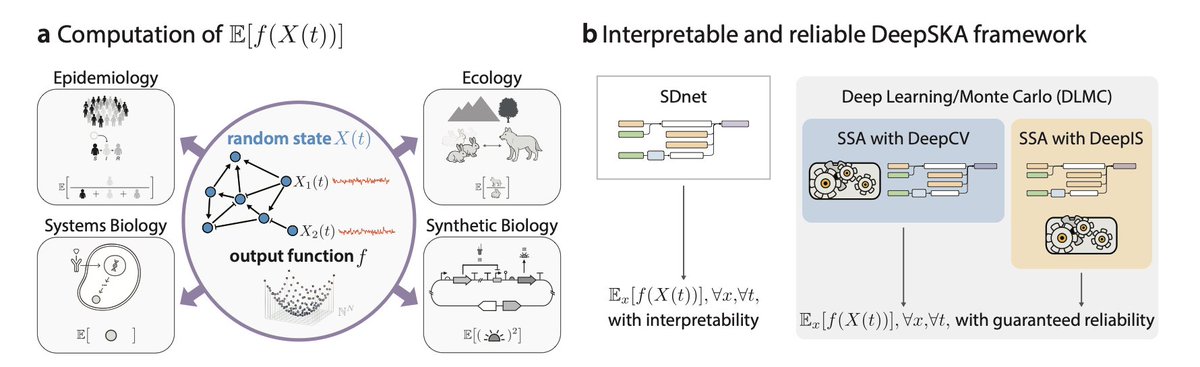
Interpretable Neural Approximation of Stochastic Reaction Dynamics with Guaranteed Reliability
1. The paper introduces DeepSKA, a novel framework combining deep learning with stochastic reaction networks (SRNs) to predict expected outputs with guaranteed reliability. This is a significant step forward in addressing the computational challenges of SRNs, which are fundamental models in systems biology, epidemiology, and ecology.
2. DeepSKA integrates a Spectral Decomposition-based network (SDnet) that provides an interpretable representation of expected outputs. Unlike traditional black-box neural networks, SDnet's structure is grounded in the spectral decomposition of SRNs, making it transparent and mathematically interpretable.
3. The framework also includes Deep Learning/Monte Carlo (DLMC) estimators that combine neural approximations with stochastic simulations. These estimators offer unbiased estimates with dramatically reduced variance compared to conventional Monte Carlo methods, achieving several orders of magnitude efficiency improvements.
4. DeepSKA demonstrates its effectiveness across nine diverse SRNs, including nonlinear and non-mass-action models with up to ten species. The framework shows strong predictive accuracy and generalization capabilities, even beyond the training horizon.
5. The study extends the framework to compute steady-state means and variances, further enhancing its applicability. The Ergodic Mean with Deep Control Variates (EM with DeepCV) and Deep Integral Path Algorithm (DeepIPA) provide reliable and efficient estimations of these quantities.
6. The authors propose leveraging decay modes from the Stochastic Koopman Approximation (SKA) as an informed initialization for DeepSKA, potentially accelerating convergence. This integration highlights the synergy between deep learning and established analytical methods.
7. The work opens up future possibilities for extending DeepSKA to other Markovian models, such as stochastic differential equations, and for exploring extrinsic variability and parameter sensitivity in biological systems.
📜Paper: arxiv.org/abs/2512.06294v1
#StochasticReactionNetworks #DeepLearning #ComputationalBiology #InterpretableAI #StochasticModeling
3
772
2 Dec 2025
VCWorld: A Biological World Model for Virtual Cell Simulation
1. VCWorld introduces a novel cell-level white-box simulator that integrates structured biological knowledge with large language models (LLMs) to predict cellular responses to perturbations, offering a transparent and interpretable alternative to traditional black-box models.
2. The core innovation lies in its biological world model, which leverages signaling pathways, protein-protein interactions, and gene regulatory networks to enhance data efficiency and generalization, even with limited training data.
3. VCWorld achieves state-of-the-art performance in predicting differential expression and directional changes in gene expression, outperforming existing models while providing mechanistic explanations aligned with biological principles.
4. The study introduces GeneTAK, a new benchmark derived from the Tahoe-100M dataset, reframing cell-drug observations into gene-centric perturbation responses to enable more granular modeling of drug effects.
5. VCWorld's reasoning process is grounded in a comprehensive biological knowledge graph, ensuring that predictions are not only accurate but also verifiable through step-by-step mechanistic hypotheses.
6. The model demonstrates robustness in few-shot learning scenarios, making it particularly valuable for predicting responses to novel perturbations not present in the training data.
7. VCWorld's design emphasizes interpretability, allowing biologists to trace the reasoning behind each prediction, which is crucial for scientific discovery and experimental design.
📜Paper: arxiv.org/abs/2512.00306v1
#VirtualCellModeling #BiologicalWorldModel #LLMs #PerturbationPrediction #InterpretableAI #ComputationalBiology

1
8
18
1,548
19 Nov 2025
Genomic Perception Fusion: A Lightweight, Interpretable Kernel for Protein Functional Tuning
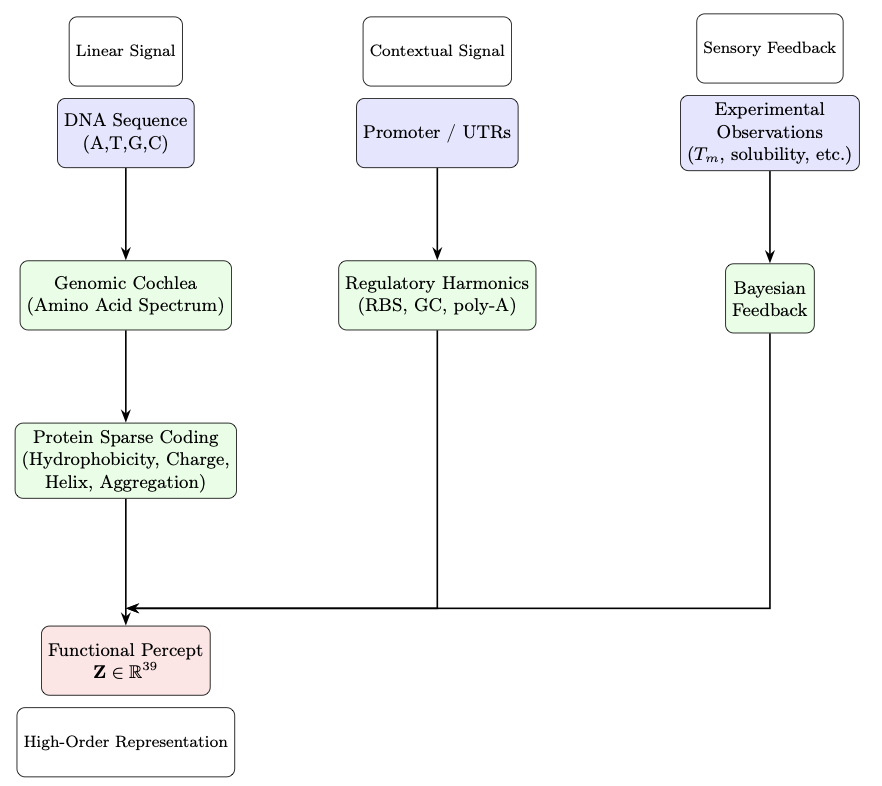
1. This new study introduces Genomic Perception Fusion (GPF), a groundbreaking algorithm that transforms protein design by treating DNA sequences as biophysical signals rather than inert code. It offers a transparent and computationally efficient alternative to deep generative models for protein engineering.
2. GPF integrates sequence composition, regulatory context, and experimental feedback into a 39-dimensional functional representation. It leverages biophysical principles to predict protein stability, solubility, and expression without requiring extensive computational resources or training on private datasets.
3. The core innovation of GPF lies in its hierarchical pipeline, which includes the Genomic Cochlea for amino acid composition analysis, Protein Sparse Coding for biophysical feature extraction, and Regulatory Harmonics for expression context modeling. This approach allows for rapid and interpretable predictions.
4. Validated against GFP surface mutations, GPF demonstrates high accuracy in forecasting the effects of mutations on protein stability and solubility. Its Bayesian updating mechanism further enhances prediction reliability by incorporating sparse experimental data.
5. Unlike black-box models, GPF provides full interpretability of its predictions, making it an ideal tool for experimental protein engineers who need fast, transparent guidance. It is best suited for optimizing existing protein scaffolds and complements generative design methods.
6. The study highlights the value of physics-informed approaches in the era of large-scale generative models. GPF runs on standard hardware in milliseconds, proving that lightweight and interpretable methods can still deliver robust results for protein engineering applications.
📜Paper: biorxiv.org/content/10.1101/…
#ProteinEngineering #ComputationalBiology #GenomicPerceptionFusion #InterpretableAI
1
7
1,069
27 Oct 2025
MolGraphormer: An Interpretable GNN-Transformer for Uncertainty-Aware Molecular Toxicity Prediction
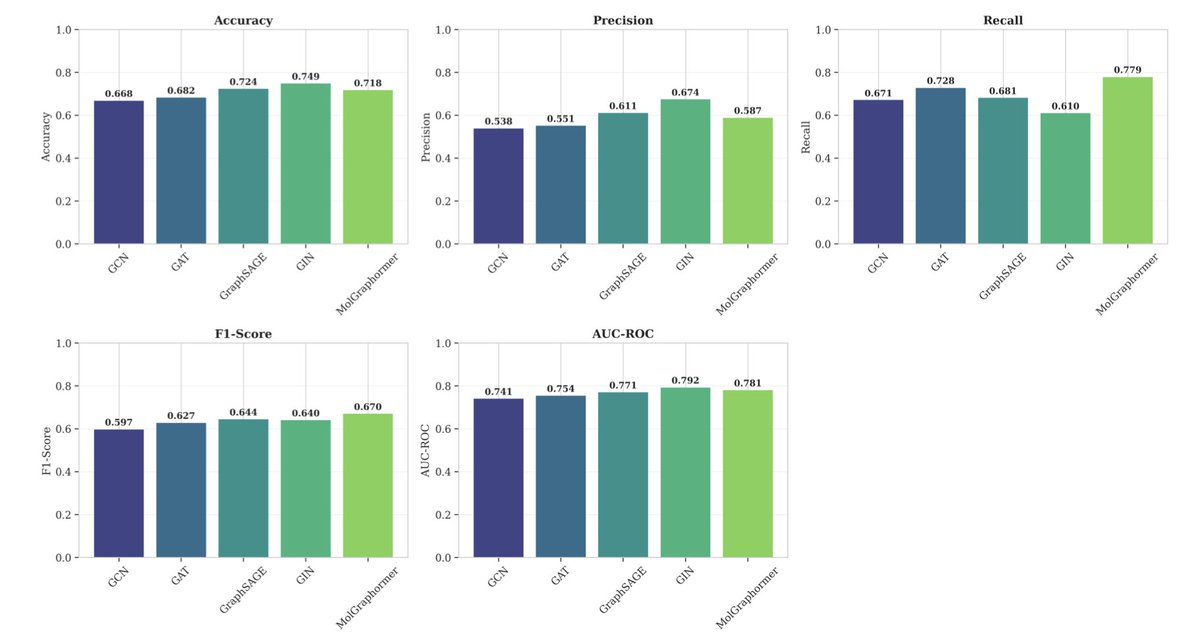
1. MolGraphormer introduces a novel hybrid architecture combining Graph Neural Networks (GNNs) with Transformer self-attention mechanisms for predicting molecular toxicity. This innovative approach integrates substructure-aware embeddings, edge-conditioned message passing, and hierarchical graph aggregation to enable both local and global molecular reasoning.
2. Evaluated on the Tox21 benchmark dataset, MolGraphormer achieves a competitive F1-score of 0.6697 and AUC-ROC of 0.7806. Notably, it maintains a strong recall of 0.7787 for identifying toxic compounds, which is crucial for safety-critical toxicity screening applications.
3. The model incorporates comprehensive uncertainty quantification using Monte Carlo Dropout and Temperature Scaling. This allows MolGraphormer to provide not only predictions but also a measure of confidence in those predictions, enhancing its reliability for drug safety assessment.
4. MolGraphormer’s attention-based interpretability reveals patterns that correlate with known toxicophores, such as high attention on aromatic rings, heteroatoms, and specific toxicophores like nitro groups and epoxides. This suggests the model learns chemically meaningful representations.
5. The architecture includes an embedding layer, multi-head graph attention layers with edge conditioning, graph pooling, and a classification head. This design enables the model to capture both local bonding and long-range structural information effectively.
6. Training dynamics show that MolGraphormer requires more epochs to converge compared to other GNN architectures due to its deeper structure with residual connections. However, this results in better final performance with minimal overfitting.
7. Despite its strengths, MolGraphormer has some limitations. It exhibits a moderate precision, resulting in a 41.3% false positive rate. Additionally, its performance degrades on rare structural scaffolds and molecules exceeding 100 atoms show increased prediction variance.
8. Future work could explore self-supervised pre-training on large molecular databases, multi-task learning across all Tox21 endpoints, integration of 3D conformational information, ensemble methods, mechanistic integration via knowledge graphs, and active learning using uncertainty.
📜Paper: doi.org/10.26434/chemrxiv-20…
#MolGraphormer #GNN #Transformer #MolecularToxicity #DrugDiscovery #InterpretableAI #UncertaintyQuantification
1
2
12
1,402