
24 Jun 2025
Leveraging neural network interatomic potentials for a foundation model of chemistry
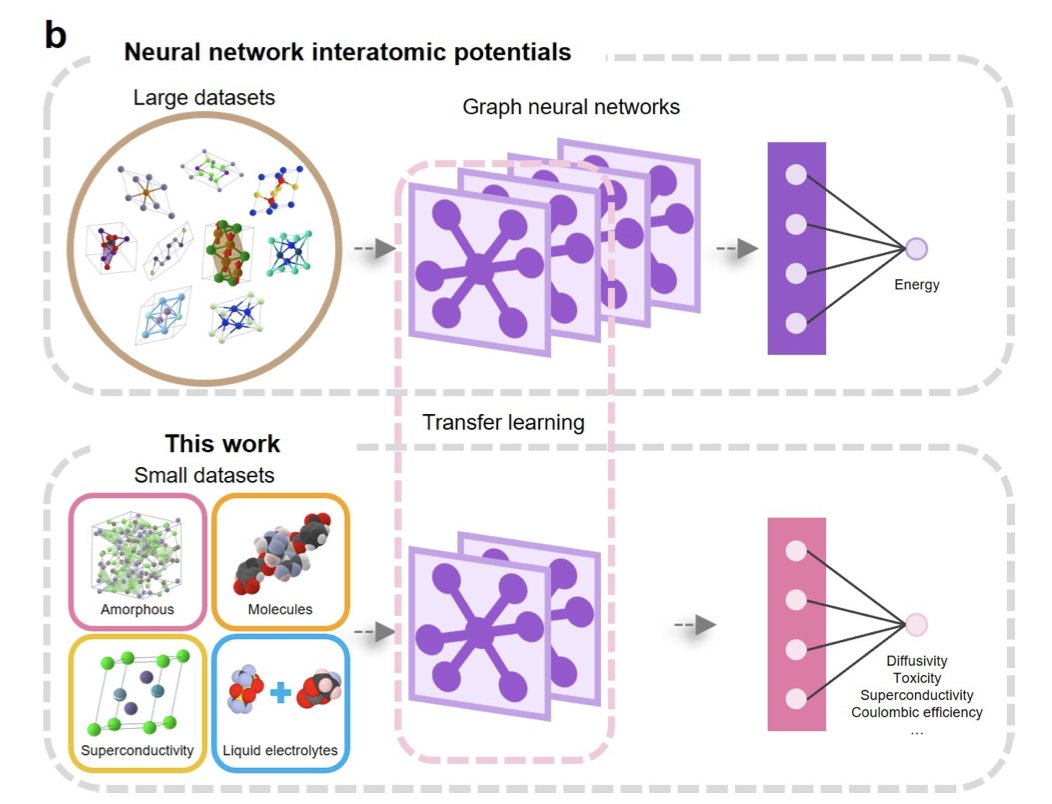
1.HackNIP is a hybrid framework combining pretrained neural interatomic potentials (NIPs) with shallow machine learning models. Instead of end-to-end deep learning, it extracts fixed-length embeddings from NIPs and feeds them into lightweight predictors for structure-to-property tasks.
2.This approach achieves state-of-the-art performance on 8 structure-property prediction tasks in the Matbench benchmark, outperforming both feature-based and deep neural network baselines in several cases.
3.HackNIP demonstrates exceptional data efficiency. On datasets with fewer than ~10⁴ samples, it often outperforms direct fine-tuning of the same NIP, making it ideal for limited-data regimes common in materials science.
4.The model remains competitive with larger end-to-end neural networks even at full dataset scale. On Matbench tasks with over 10⁴ samples, HackNIP shows performance within chemical accuracy and Pareto-optimal error margins.
5.HackNIP generalizes well beyond DFT-predicted properties. It achieves strong performance on diverse experimental and ab initio datasets, including Li-ion diffusivity, superconducting transition temperatures, and battery Coulombic efficiency.
6.It also shows robust classification performance on molecular-property prediction tasks from MoleculeNet (e.g., BBB penetration, toxicity), with AUC scores comparable to state-of-the-art molecular pretraining models despite using solid-material-trained NIPs.
7.A key innovation is identifying which NIP layer embeddings provide optimal features. Early layers often yield better predictions than deeper ones, especially for solid-state tasks, while molecular tasks benefit from deeper layers.
8.UMAP visualization and Jensen–Shannon divergence analyses show that early embeddings retain more task-relevant diversity. Late-layer embeddings become too compressed, losing fine-grained structural information crucial for downstream tasks.
9.Compared to end-to-end fine-tuning, HackNIP's simpler regressors undergo more uniform and efficient parameter updates, with less overfitting and better transferability, especially in small data regimes.
10.This modularity allows users to plug-and-play different NIP backbones and shallow learners. ORB MODNet yields the best results overall, but other combinations (e.g., MACE XGBoost) can be effective under different settings.
11.HackNIP lowers the entry barrier to high-performance materials ML. It is lightweight enough to run on standard hardware, robust across datasets, and suitable for practical use cases in both academia and industry.
12.This work suggests that hybrid approaches—assigning distinct roles to feature extraction and property mapping—can outperform monolithic models while improving interpretability and reducing computational cost.
💻Code: github.com/parkyjmit/HackNIP
📜Paper: arxiv.org/abs/2506.18497v1
#MachineLearning #MaterialsScience #TransferLearning #InteratomicPotentials #ML4Materials #ComputationalMaterials #GraphNeuralNetworks #NLPforScience
1
516
24 Jun 2025
Leveraging neural network interatomic potentials for a foundation model of chemistry
1.HackNIP is a hybrid framework combining pretrained neural interatomic potentials (NIPs) with shallow machine learning models. Instead of end-to-end deep learning, it extracts fixed-length embeddings from NIPs and feeds them into lightweight predictors for structure-to-property tasks.
2.This approach achieves state-of-the-art performance on 8 structure-property prediction tasks in the Matbench benchmark, outperforming both feature-based and deep neural network baselines in several cases.
3.HackNIP demonstrates exceptional data efficiency. On datasets with fewer than ~10⁴ samples, it often outperforms direct fine-tuning of the same NIP, making it ideal for limited-data regimes common in materials science.
4.The model remains competitive with larger end-to-end neural networks even at full dataset scale. On Matbench tasks with over 10⁴ samples, HackNIP shows performance within chemical accuracy and Pareto-optimal error margins.
5.HackNIP generalizes well beyond DFT-predicted properties. It achieves strong performance on diverse experimental and ab initio datasets, including Li-ion diffusivity, superconducting transition temperatures, and battery Coulombic efficiency.
6.It also shows robust classification performance on molecular-property prediction tasks from MoleculeNet (e.g., BBB penetration, toxicity), with AUC scores comparable to state-of-the-art molecular pretraining models despite using solid-material-trained NIPs.
7.A key innovation is identifying which NIP layer embeddings provide optimal features. Early layers often yield better predictions than deeper ones, especially for solid-state tasks, while molecular tasks benefit from deeper layers.
8.UMAP visualization and Jensen–Shannon divergence analyses show that early embeddings retain more task-relevant diversity. Late-layer embeddings become too compressed, losing fine-grained structural information crucial for downstream tasks.
9.Compared to end-to-end fine-tuning, HackNIP's simpler regressors undergo more uniform and efficient parameter updates, with less overfitting and better transferability, especially in small data regimes.
10.This modularity allows users to plug-and-play different NIP backbones and shallow learners. ORB MODNet yields the best results overall, but other combinations (e.g., MACE XGBoost) can be effective under different settings.
11.HackNIP lowers the entry barrier to high-performance materials ML. It is lightweight enough to run on standard hardware, robust across datasets, and suitable for practical use cases in both academia and industry.
12.This work suggests that hybrid approaches—assigning distinct roles to feature extraction and property mapping—can outperform monolithic models while improving interpretability and reducing computational cost.
💻Code: github.com/parkyjmit/HackNIP
📜Paper: arxiv.org/abs/2506.18497v1
#MachineLearning #MaterialsScience #TransferLearning #InteratomicPotentials #ML4Materials #ComputationalMaterials #GraphNeuralNetworks #NLPforScience

5
804